Rétinoblastome
Le rétinoblastome est une tumeur maligne de la rétine, rare (un enfant sur 15 à 20 000 serait touché) et d'origine génétique, apparaissant habituellement avant l'âge de 5 ans.
| Médicament | (RS)-cyclophosphamide |
|---|---|
| Spécialité | Oncologie |
| CIM-10 | C69.2 |
|---|---|
| CIM-9 | 190.5 |
| ICD-O | M9510/3 |
| OMIM | 180200 |
| DiseasesDB | 11434 |
| MedlinePlus | 001030 |
| eMedicine | 1222849 |
| MeSH | D012175 |
| GeneReviews | Retinoblastoma |
| Patient UK | Retinoblastoma-pro |
![]() Mise en garde médicale
Mise en garde médicale
| Rétinoblastome | |
| Référence MIM | 180200 |
|---|---|
| Transmission | Récessive, mais la deuxième mutation a lieu dans 97 % des cas |
| Chromosome | 13q14.1-q14.2 |
| Gène | RB1 |
| Mutation | Ponctuelle |
| Mutation de novo | Rare |
| Nombre d'allèles pathologiques | Plus de 400 |
| Incidence | 1 pour 20 000 naissances |
| Pénétrance | Variable |
| Maladie génétiquement liée | Ostéosarcome |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
Épidémiologie
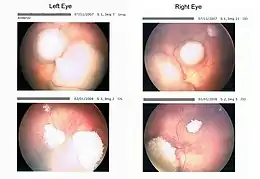
Le rétinoblastome peut atteindre un œil ou les deux. La moitié des rétinoblastomes est diagnostiquée avant l'âge de deux ans et 45 % touchent les deux yeux avant l'âge de 15 mois. L'incidence varie, suivant les études, entre 3 et 50 par million d'enfants de moins de 14 ans[1].
Génétique
Ce cancer survient chez les enfants porteurs de deux allèles pathologiques du gène RB1 situé sur le chromosome 13.
Une cause génétique a été suspectée dès les années 1970[2]. Le développement du cancer est dû à une mutation présente sur chacun des deux chromosomes 13 en position 13q14. Elle a été découverte dans les années 1980[3]. La protéine codée, Rb, est alors non fonctionnelle. Son rôle est le contrôle négatif du cycle cellulaire, c'est pourquoi si elle est mutée la cellule se divise sans frein, causant ainsi la tumeur. La protéine Rb est présente dans toutes les cellules du corps, mais c'est dans la rétine qu'elle a le rôle le plus important car il y a peu d'autres freins au cycle cellulaire à cet endroit. L'inactivation isolée de la protéine Rb est cependant nécessaire mais pas suffisante pour amorcer le processus tumoral[4]. Elle provoquerait une « instabilité » du génome[5] expliquant la cancérisation. Cette inactivation peut être (rarement) partielle conduisant à une pénétrance réduite[6].
Les porteurs d'un seul allèle pathologique auront un risque augmenté de cancer non oculaire en rapport avec le gène RB. La survenue d'un deuxième cancer (ostéosarcome dans la moitié des cas) est fréquente : le risque sur 30 ans varie de 35 % (s'il y a eu une radiothérapie contre le rétinoblastome) à 60 % (s'il n'y en a pas eu). Bien qu'en théorie la radiothérapie augmente le risque de développer un cancer plusieurs années après le traitement, dans ce cas-là, c'est l'inactivation de RB1 qui est à l'origine de la seconde néoplasie.
La mutation peut être « de novo », et donc, non héritée du père ou de la mère, ou transmise par ces derniers. Dans ce cas, la mutation vient le plus souvent d'une cellule germinale du père[7]. Dans de rares cas, la mutation peut être de type mosaïque[8], c'est-à-dire, présente seulement sur certaines cellules. Les rétinoblastomes bilatéraux sont héréditaires. Les formes unilatérales ne sont pas transmissibles aux enfants dans la majorité des cas[9].
Diagnostic
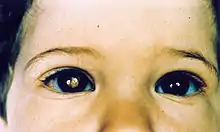
Deux signes connus indiquent un risque de rétinoblastome chez une personne :
- une leucocorie (reflet blanchâtre de la pupille, apparaissant sous un certain angle, et parfois mis en évidence sur des photos prises au flash[10])
- un strabisme.
Le diagnostic de rétinoblastome est porté par un ophtalmologiste. Celui-ci réalise un examen du fond d’œil avec dilatation de la pupille et, en cas de suspicion de rétinoblastome, adresse l'enfant vers un centre spécialisé qui réalisera un nouveau fond d’œil dilaté et un examen de la rétine, le plus souvent sous anesthésie générale.
L'examen ophtalmologique peut être complété par une IRM et une échographie oculaire. En cas de tumeur très volumineuse, d'envahissement rétinien ou du nerf optique, la recherche de métastases à distance peut être nécessaire[11].
Traitement
La prise en charge du rétinoblastome a fait l'objet de la publication de recommandations par la Canadian Retinoblastoma Society[12].
Les traitements sont soit la chirurgie avec parfois une ablation chirurgicale de l'œil, la chimiothérapie (cisplatine, étoposide et vincristine[13]), le laser diode, la thermo-chimiothérapie, la cryothérapie, la radiothérapie ou la photocoagulation.
L'énucléation est souvent la seule option dans les formes non diffuses dans les pays en voie de développement. L'examen anatomopathologique de l'œil permet de voir si la tumeur est restée intra-oculaire.
La radiothérapie externe a été utilisée dès les années 1950[14] mais son utilisation augmente le risque de survenue d'un second cancer dans les années qui suivent[15]. Une radiothérapie plus ciblée, dite stéréotaxique, pourrait être un traitement d'appoint dans certaines formes[16].
Un contrôle régulier du fond d'œil et un suivi pédiatrique sont nécessaires.
Le comédien américain Peter Falk , célèbre notamment pour son rôle du lieutenant Columbo, a été atteint d'un rétinoblastome à l'âge de 3 ans, et a porté un œil de verre depuis cet âge et jusqu'à la fin de sa vie.
L’actrice française Claude Jade meurt de métastases hépatiques causées par ce cancer de l’œil le .
Association
En France, RETINOSTOP[17] est l'association française de lutte contre le rétinoblastome.
Notes et références
- (en) Kivela T, « The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death » Br J Ophthalmol. 2009;93:1129-1131.
- (en) Knudson AG, « Mutation and cancer: statistical study of retinoblastoma » Proc Natl Acad Sci U S A. 1971;68:820-823
- (en) Friend SH, Bernards R, Rogelj S et al. « A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma » Nature. 1986;323:643-646.
- (en) Corson TW, Gallie BL, « One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma » Genes Chromosomes Cancer. 2007;46:617-634.
- (en) Dimaras H, Khetan V, Halliday W et al. « Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma » Hum Mol Genet. 2008;17:1363-1372.
- (en) Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B, « Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma » Hum Genet. 1994;94:349-354.
- (en) Zhu XP, Dunn JM, Phillips R et al. « Preferential germline mutation of the paternal allele in retinoblastoma » Nature. 1989;340:312-313.
- (en) Rushlow D, Piovesan B, Zhang K et al. « Detection of mosaic RB1 mutations in families with retinoblastoma » Hum Mutat. 2009;30:842-851
- (en) Dimaras H, Kimani K, Dimba EA et al. « Retinoblastoma » Lancet. 2012;379:1436 - 1446.
- Gaubert C (2019) Cancer de l’œil : une petite fille sauvée par une photo montrant un étrange reflet blanc, article publié par Science et Avenir le 19.04.2019
- « Rétinoblastome : comment établit-on le diagnostic ? | Institut Curie », sur curie.fr (consulté le )
- Canadian Retinoblastoma Society. « National Retinoblastoma Strategy Canadian guidelines for care ; stratégie thérapeutique du rétinoblastome guide clinique canadien » Can J Ophthalmol. 2009;44(suppl 2):S1-88.
- (en) Chan HS, Gallie BL, Munier FL, Beck Popovic M, « Chemotherapy for retinoblastoma » Ophthalmol Clin North Am. 2005;18:55-63.
- (en) Stallard HB, « Irradiation of retinoblastoma (glioma retinæ) », Lancet. 1952;1:1046-1049.
- (en) Eng C, Li FP, Abramson DH et al. « Mortality from second tumors among long-term survivors of retinoblastoma » J Natl Cancer Inst. 1993;85:1121-1128.
- (en) Sahgal A, Millar BA, Michaels H et al. « Focal stereotactic external beam radiotherapy as a vision-sparing method for the treatment of peripapillary and perimacular retinoblastoma: preliminary results » Clin Oncol (R Coll Radiol). 2006;18:628-634.
- Association française de lutte contre le rétinoblastome
Voir aussi
Articles connexes
Bibliographie
- (en) Pathologic Basis of Disease, Robbins and Cotran & The Cell, Alberts
Liens externes
- Notices dans des dictionnaires ou encyclopédies généralistes :
- Ressources relatives à la santé :
- Classification internationale des maladies oncologiques
- GeneReviews
- ICD9Data.com
- Orphanet
- (en) Diseases Ontology
- (en) DiseasesDB
- (en + es) Genetic and Rare Diseases Information Center
- (en) Héritage mendélien chez l'humain
- (en) Héritage mendélien chez l'humain
- (en) ICD-10 Version:2016
- (en) Medical Subject Headings
- (en + es) MedlinePlus
- (en) NCI Thesaurus
- (cs + sk) WikiSkripta
- (en) Medical Genetics Information Resource