Leucémie myéloïde chronique
La leucémie myéloïde chronique (LMC) est une prolifération myéloïde monoclonale sans blocage de maturation prédominant sur la lignée granuleuse au niveau médullaire et splénique.
| Médicament | Uramustine, busulfan, idarubicine, (RS)-cyclophosphamide, plicamycin (en), hydroxyurée, hydrate de thioguanine (d), mésilate d'imatinib (d) et pipobroman |
|---|---|
| Spécialité | Oncologie |
| CIM-10 | C92.1 |
|---|---|
| CIM-9 | 205.1 |
| ICD-O | M9875/3 |
| OMIM | 608232 |
| DiseasesDB | 2659 |
| MedlinePlus | 000570 |
| eMedicine | 199425 |
| MeSH | D015464 |
![]() Mise en garde médicale
Mise en garde médicale
Dans l'espèce humaine, elle fait partie des 4 grands syndromes myéloprolifératifs (avec la maladie de Vaquez, la thrombocytémie essentielle et la splénomégalie myéloïde). Elle touche surtout l'adulte entre 30 et 50 ans et est favorisée par l'exposition au benzène et aux rayons ionisants.
Symptômes cliniques
Le début est insidieux et certains cas sont découverts fortuitement lors d'un hémogramme de routine chez des sujets apparemment bien portants.
Symptômes généraux
L'état général est bien conservé au début. La fièvre est absente ou se réduit à des fébricules. Pourtant le patient éprouve de la fatigue, des sueurs, de l'inappétence, parfois de la dyspnée. Certains sujets peuvent au début être suspectés d'hyperthyroïdie : on trouve en effet un métabolisme de base accru, mais c'est par suite de la prolifération leucémique (hypermétabolisme de tous les états néoplasiques).
Symptômes en rapport avec l'anomalie sanguine
L'anémie et la tendance aux infections manquent généralement durant les premiers mois de l'évolution car les globules blancs sont fonctionnels. Un peu plus souvent on note une tendance aux hémorragies (hémorragies gingivales, épistaxis, purpura, hémorragies à l'occasion d'avulsions dentaires).
Symptômes en rapport avec le développement du tissu leucémique
a) La rate est grande (splénomégalie) : elle atteint parfois la fosse iliaque gauche. Elle est dure et indolore sauf s'il existe de la périsplénite. Dans ce cas on peut percevoir un frottement au palper ou à l'auscultation. La splénomégalie se manifeste souvent par une pesanteur à l'épigastre ou à l'hypochondre gauche, parfois par de vives douleurs en cas de périsplénite.
b) Les ganglions lymphatiques ne sont pas hypertrophiés dans la majorité des cas. Les formes avec adénopathies sont généralement considérées comme graves.
c) Le foie est souvent un peu augmenté de volume, du moins à la fin.
d) Les os peuvent être douloureux, spontanément ou à la pression.
Cytogénétique et étiologie
Anomalies initiales
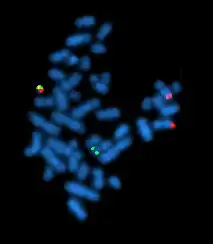
- Chromosome de Philadelphie (Ph) :
c’est un chromosome 22 raccourci sur son bras long, résultat le plus souvent d’une translocation réciproque t(9;22)(q34;q11) dite standard. Il est présent, sous cette forme ou d’autres, dans plus de 90 % des cas. - Points de cassure géniques :
- situé sur le chromosome 9 (où on note un gain de longueur sur le bras long), dans le grand intron 1 du gène ABL qui, par son produit, nucléaire et cytoplasmique, contrôle, avec ATM, RB1 et p53, la multiplication cellulaire, notamment si des lésions de l’ADN requièrent réparation ;
- et sur le chromosome 22, entre les exons b2 et b3 ou b3 et b4, dans les quelques kb de la zone M-bcr du gène BCR, où les points d’impact de toutes les malades sont groupés.
- Le néogène BCR/ABL :
Un néogène chimérique se forme : BCR/ABL. La partie initiale de BCR, avec son promoteur, restée sur le chromosome 22, est mise en continuité avec le fragment d'ABL, représentant la presque totalité du gène, venu du chromosome 9.
Le messager est traduit en une protéine hybride, p210 Bcr-Abl (de 210 kDa). Elle domine largement, associée à une faible quantité d’une forme plus courte, p190, provenant ici d’un épissage alternatif du messager (isolée, elle caractérise les leucémies aigües lymphoblastiques). - Variantes
Les translocations complexes (un peu moins de 10 % des cas) impliquent un ou plusieurs chromosomes en plus du chromosome 9 et du chromosome 22, mais ont toujours pour résultat la production de protéines b2/a2 ou b3/a2.
Chez 5 % des patients, BCR/ABL peut être mis en évidence alors que le caryotype est normal. Dans ces LMC Ph- BCR/ABL+, d’aspect et d’évolution habituels, c’est le seul marqueur de la population maligne et on doit recourir à la biologie moléculaire (et/ou l’hybridation in situ) pour identifier ces cas et les suivre.
L'influence respective de ces diverses espèces protéiques reste controversée. Il existe de rares formes, à p230, dites à polynucléaires, avec souvent aussi une hyperplaquettose d’évolution plus lente et une entité myélomonocytaire, rare, agressive, à p190. - LMC dites atypiques
Environ 5 % des cas évoquant avant tout au départ des LMC n’ont ni Ph ni fusion BCR/ABL. Des nuances hématologiques distinguent ces hémopathies, d’évolution plus défavorable.
Anomalies secondaires
Des anomalies chromosomiques additionnelles (chromosomes surnuméraires + 8, + Ph, + 19, présence d’un iso 17q) et/ou géniques (frappant p53, p16, Rb) sont quasi constantes au moment de la TA. Elles résultent d’une instabilité génomique, qui dépend de Bcr-Abl et est partie intégrante de la LMC. Elle pourrait être liée, au moins en partie, à une méthylation anormale de divers sites de l’ADN. Le nombre et la nature de ces anomalies influencent le pronostic, le choix thérapeutique et l'évolution de la maladie.
Examens de laboratoire
- Hémogramme
- Les globules blancs sont supérieurs à 50 G/L et classiquement entre 100 et 300 G/L (soit 100 000 à 300 000 / mm³). Leur nombre peut toutefois être beaucoup plus élevé, et atteindre le 1000 G/L (soit 1 million / mm3) : c'est dans ces cas, où la sédimentation du sang en tube montre une épaisse couche crémeuse leucocytaire entre le sédiment érythrocytaire et la couche de plasma, que la maladie mérite vraiment le nom de leucémie (« sang blanc ») que lui a donné Virchow. Les formes sub- et aleucémiques de la myélose chronique sont rares. Du point de vue qualitatif, ces G.B. sont essentiellement représentés par des granulocytes mûrs, principalement des neutrophiles (50-70 %). Les éosinophiles et les basophiles sont également très nombreux. Le reste de la population blanche du sang est représenté par les précurseurs immédiats des granulocytes : métamyélocytes, myélocytes et promyélocytes, surtout neutrophiles. Les myéloblastes sont peu nombreux (1-5 %). Leur apparition en nombre signe le début de la transformation aiguë.
- Globules rouges : l'anémie est nulle ou modérée, du moins au début. On peut trouver quelques érythroblastes. À la période terminale l'anémie devient grave.
- Plaquettes : au début le nombre de plaquettes est fréquemment augmenté. La thrombopénie s'installe avec la transformation aiguë.
- Myélogramme
- La moelle osseuse est très riche en cellules, elle montre une hyperplasie du tissu granulopoiétique.
- Cytogénétique et biologie moléculaire
- Caryotype : détection et suivi du chromosome de Philadelphie t(9:22) et d'éventuelles anomalies chromosomiques additionnelles. 95 % des cas sont Ph+, seuls 5 % sont Ph-.
- FISH : recherche et suivi du gène de fusion Bcr-Abl.
- RT-PCR
- Autres examens : le taux d'acide urique du sang et l'excrétion urinaire d'acide urique sont généralement augmentés, en rapport avec l'hypermétabolisme des acides nucléiques du tissu en prolifération.
Évolution et pronostic
La leucémie myéloïde chronique (LMC) est longtemps restée une maladie de mauvais pronostic, se transformant inévitablement en leucémie aiguë pouvant échapper à toute possibilité thérapeutique.
La greffe allogénique de moelle osseuse était alors le seul traitement curatif disponible, mais tous les patients ne pouvaient pas en bénéficier, les donneurs compatibles étant rares et son taux de mortalité approchant les 20 %.
Depuis le début des années 2000, des nouvelles thérapies dites ciblées, permettant de freiner la croissance anarchique des cellules cancéreuses, ont transformé le pronostic de la maladie. Les patients atteints de LMC peuvent désormais espérer voir leur état se stabiliser au point de permettre la poursuite d'une activité professionnelle, sous réserve d'un traitement continu, avec une espérance de vie normale.
Le premier représentant de ces nouvelles thérapies est l'imatinib mésylate (Glivec), inhibiteur compétitif de l'activité tyrosine-kinase BCR/ABL. Il a reçu en 2001 l'autorisation européenne de mise sur le marché en tant que médicament orphelin pour le traitement de la leucémie myéloïde chronique.
De nouvelles molécules sont disponibles pour les sujets résistants au Glivec, telles que le dasatinib (Sprycel) commercialisé depuis 2006 et le nilotinib (Tasigna) depuis 2008.
Diagnostics différentiels
La LMC ne peut guère être confondue qu'avec les réactions leucémoïdes qui sont des états non-leucémiques dans lesquels le tableau hématologique et parfois le tableau clinique (anémie, grosse rate, hémorragies) présentent une certaine ressemblance avec les leucémies. Le diagnostic différentiel peut être éventuellement une thrombocytémie essentielle. Le caryotype permet de différencier totalement la pathologie.
Complications
- Vasculaires : thromboses et hémorragies
La thrombocytose et la thrombopénie qui accompagne tout syndrome myéloprolifératif peut être la cause de thromboses veineuses et d'hémorragies (penser à l'ulcère gastroduodénal). La thrombopathie qui peut être associée majore le risque hémorragique. - Vasculaires : leucostase
La leucostase due à l'hyperleucocytose peut provoquer une insuffisance respiratoire aiguë. Au fond d'œil, on peut également observer une rétinite leucémique. - Métaboliques : hyperuricémie
L'hyperuricémie, conséquence de l'hyperleucocytose, peut se manifester par des crises de goutte ou des coliques néphrétiques - Hématologique : splénomégalie
La splénomégalie peut se compliquer d'une hémodilution et d'un hypersplénisme s'accompagnant d'une thrombopénie et d'une anémie. Lorsqu'elle est majeure, il peut y avoir des infarctus spléniques voire un risque de rupture splénique. - Hématologique : insuffisance médullaire
La myélofibrose qui accompagne la maladie peut être la cause d'une insuffisance médullaire. On retrouve alors une pancytopénie (anémie, thrombopénie et neutropénie) se manifestant par une asthénie, des hémorragies et des infections à pyogènes. L'insuffisance médullaire peut également être due à un envahissement médullaire par des cellules immatures, les blastes, au cours de la phase accélérée ou aiguë de la maladie. - Hématologique : leucémie aiguë
La transformation en leucémie aiguë secondaire ou acutisation est constante lors de l'évolution non traitée. Dans 70 % des cas, il s'agit d'une leucémie aiguë myéloïde, dans 20 % d'une leucémie aiguë lymphoïde et dans 10 % d'une leucémie aiguë indifférenciée. La transformation se manifeste par une altération de l'état général, l'apparition d'un syndrome tumoral, d'une insuffisance médullaire et par une augmentation des blastes sur l'hémogramme. De très mauvais pronostic, l'évolution de cette leucémie aiguë secondaire est habituellement fatale en moins de 6 mois. Les traitements actuels, greffe de moelle, interféron, Glivec ont transformé le pronostic.
Traitement
À de très rares exceptions près, la leucémie myéloïde chronique reste incurable. Avant l'arrivée récente des thérapies ciblées, la médiane de survie, avec les médicaments classiques comme le Misulban et l'Hydréa était de 5 ans et l'issue était constamment fatale par transformation en leucémie aiguë. Le traitement visait à retarder au maximum la transformation aiguë et à contrôler l'hyperleucocytose pour prévenir les complications.
Une guérison est possible après transplantation de moelle osseuse soit familiale (fratrie) soit donneur non familial pour les patients de moins de 50 ans[1].
L'interféron alpha obtient des rémissions beaucoup plus longues.
Plusieurs révolutions médicamenteuses sont également en cours, avec l'apparition, depuis la fin des années 1990, sur le marché de nouveaux médicaments: ainsi le Glivec, le Sprycel, ou bien encore le Tasigna. En ce qui concerne le Glivec (Imatinib) il transforme radicalement le pronostic, en permettant aux patients, sous réserve d'un bon suivi du traitement, d'avoir une espérance de vie normale, et une bonne qualité de vie. Ce traitement mis sur le marché en 2001 permet en effet de cibler les cellules cancéreuses: l'anomalie chromosomique de la LMC provoque une translocation entre les chromosomes 9 et 22 (au niveau des gènes bcr et abl) qui permet la formation d'une protéine qui possède une activité tyrosine kinase aberrante (exacerbée). Or, c'est cette activité qui « donne l'ordre » aux cellules de proliférer de façon anarchique et les empêche de subir l'apoptose (mort cellulaire programmée). Le Glivec a une action inhibitrice de l'activité tyrosine kinase de bcr-abl car il va se fixer sur le site de liaison de l'adénosine triphosphate de la protéine, l'empêchant de fonctionner correctement et de donner ses ordres de prolifération cancéreuse. C'est donc un traitement ciblé à ce jour qui est maintenant utilisé en 1re intention.
Le Glivec est un médicament très cher (2 309,69€ en France, en 2014, la boîte de trente comprimés de Glivec 400). S'il est remboursé à 100 % par la Sécurité Sociale en France et depuis 2002 en Belgique[2], beaucoup de malades des pays pauvres ne peuvent y avoir recours. Cette situation entraîne les protestations de nombreuses associations humanitaires. En , la Cour suprême de New Delhi a rejeté le brevet déposé par la société Novartis afin de permettre le développement d'un médicament générique accessible au plus grand nombre, au grand dam des sociétés pharmaceutiques qui s'inquiètent des répercussions que cela pourrait avoir sur l'innovation médicale[3].
Notes et références
- « Tout savoir sur le don | Don de Moelle Osseuse », sur www.dondemoelleosseuse.fr (consulté le )
- « Le Glivec est à présent remboursé », sur La Libre Belgique, (consulté le ).
- Julien Bouissou, « Affaire Novartis : l'Inde préserve les médicaments génériques », lemonde.fr, (lire en ligne)
Voir aussi
Articles connexes
Liens externes
- CMLeukemia
- Leucémie myéloïde chronique sur "Orphanet"
- France intergroupe des Leucémies myéloïdes chroniques Association de médecins, biologistes et pharmaciens spécialistes de la LMC
- LMC France Association de patients et proches de patients touchés par la LMC (leucémie myéloïde chronique).