Syndrome d'Eisenmenger
Le syndrome d'Eisenmenger est un ensemble de symptômes traduisant la présence d'une hypertension artérielle pulmonaire « fixée » venant compliquer l'évolution spontanée des cardiopathies congénitales comportant un shunt gauche-droit responsable d'une augmentation importante et prolongée du débit sanguin dans la circulation pulmonaire.
| Spécialité | Génétique médicale |
|---|
| CIM-10 | Q21.8 |
|---|---|
| CIM-9 | 745.4 (CDC/BPA 745.410) |
| DiseasesDB | 4143 |
| MedlinePlus | 007317 |
| eMedicine | 154555 |
| MeSH | D004541 |
![]() Mise en garde médicale
Mise en garde médicale
Cette hypertension artérielle pulmonaire est la conséquence d'une maladie vasculaire touchant l'ensemble de l'arbre artériel pulmonaire et parfois désignée sous le terme d'artériolithe (« artères de pierre ») pulmonaire.
Chez l'enfant, c'est la cause principale d'hypertension artérielle pulmonaire dite « secondaire », par opposition à l'hypertension artérielle pulmonaire « primitive », beaucoup plus rare à cet âge.
Dans les pays développés, l'apparition d'un syndrome d'Eisenmenger tend à devenir exceptionnelle grâce à la prise en charge chirurgicale précoce (palliative ou curative) des cardiopathies s'accompagnant d'un shunt gauche-droit. Ce syndrome reste un fléau sévissant dans les pays en voie de développement en raison d'un retard au diagnostic ou à la prise en charge.
Une fois constitué, le syndrome d'Eisenmenger est irréversible et les traitements actuels ne visent qu'à ralentir son évolution ou combattre ses conséquences, la plus visible étant l'apparition d'une cyanose liée à l'inversion du sens du shunt causal, qui devient alors droit-gauche.
Il a été décrit par Victor Eisenmenger en 1897[1].
Mécanisme
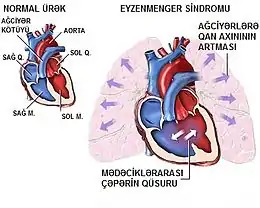
Notion de shunt gauche-droit
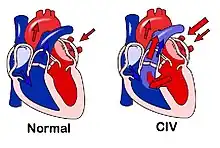
On parle de shunt (c'est-à-dire « court-circuit ») quand une partie du sang emprunte un « chemin » plus court que celui qu'il aurait dû parcourir normalement. L'expression « gauche-droit » caractérise le sens du shunt, qui se produit d'une cavité gauche (oreillette ou ventricule) ou de l'aorte vers une des structures du ventricule droit (oreillette - ventricule - circulation pulmonaire).
En raison des différences de pression régnant normalement dans les cavités cardiaques et les gros vaisseaux issus du cœur, toute communication isolée entre le cœur gauche et le cœur droit est responsable d'un shunt gauche-droit plus ou moins important. Les plus fréquentes sont :
- les communications inter-ventriculaires entre les deux ventricules ;
- les communications inter-auriculaires entre les deux oreillettes ;
- la persistance du canal artériel entre l'aorte et l'artère pulmonaire.
D'autres malformations cardiaques, plus rares, sont également responsables d'un shunt gauche-droit :
- canal atrio-ventriculaire (dans une forme partielle ou complète) ;
- fistule ou fenêtre aorto-pulmonaire ;
- communication ventricule gauche-oreillette droite ;
- fistules coronaro-cardiaques ou coronaro-pulmonaires ;
- certaines formes de Tronc artériel commun…
Surcharge de la vascularisation pulmonaire et du cœur gauche
En présence d'une communication, le sang présent dans le cœur gauche peut emprunter le parcours normal, vers l'aorte et l'ensemble du corps (« grande » circulation ou circulation « systémique »), ou passer à travers la communication dans le cœur droit pour retourner directement vers les poumons d'où il provient (« petite » circulation ou circulation « pulmonaire »).
La quantité de sang qui passe par la communication et emprunte le court-circuit est d'autant plus importante que la communication est large et que la différence de pression entre les cœurs gauche et droit est importante. Quand la communication est large, elle n'oppose pratiquement aucune restriction au passage du sang (« shunt non restrictif ») et la quantité qui passera sera uniquement fonction de la différence de pression de part et d'autre de la communication.
Les conséquences d'un shunt gauche-droit sont liées à l'augmentation de la quantité de sang qui revient aux poumons puis, de là, dans les cavités gauches. Il faut savoir qu'en présence d'une communication large, il peut passer 2, 3 voire 4 fois plus de sang par le court-circuit que par le trajet normal vers l'aorte. Cela veut dire que les poumons reçoivent 3,4 ou 5 fois plus de sang que la normale (la quantité normale + la quantité passant par le shunt). De même, à la sortie des poumons, l'oreillette puis le ventricule gauche doivent faire circuler jusqu'à 5 fois plus de sang que normalement : un nourrisson porteur d'un large shunt se trouve donc en permanence dans un état proche d'un effort maximal. Cet excès de sang au niveau des poumons peut provoquer un œdème pulmonaire et une insuffisance ventriculaire gauche.
Altérations tissulaires de la vascularisation artérielle pulmonaire
Normalement, les artères pulmonaires ont une structure tissulaire adaptée à un fonctionnement sous un faible régime de pression. En présence d'un shunt gauche-droit, elles doivent s'adapter à l'augmentation de débit. Elles le font d'abord par une vasodilatation puis, si cela ne suffit pas, par une augmentation de pression. Dans les premiers mois de vie, cette adaptation est réversible. La fermeture de la communication anormale s'accompagne d'un retour à la normale des artères pulmonaires (diamètre et pression). Si cette situation perdure, il se produit des modifications de la structure de ces vaisseaux qui les transforment de façon irréversible en artères faites pour fonctionner sous fortes pressions. L'hypertension artérielle pulmonaire (HTAP) « dynamique » et réversible du début s'est transformée en une hypertension artérielle pulmonaire «fixée» et définitive. On parle alors de syndrome d'Eisenmenger.
Il n'existe pas de différence importantes entre les anomalies tissulaires retrouvées lors d'un syndrome d'Eisenmenger ou lors d'une hypertension artérielle pulmonaire de cause autre[2].
Inversion du shunt
Lorsque la pression des cavités droites excèdent celles des cavités gauches, le shunt s'inverse, devenant alors droit-gauche, ce qui est constitutif du syndrome d'Eisenmenger. Du sang pauvre en oxygène (veineux) rejoint alors la circulation artérielle, expliquant la cyanose. Cette désaturation chronique provoque une polyglobulie réactionnelle.
Délai de constitution d'un syndrome d'Eisenmenger
Ce délai est variable. Fonction du siège de la communication entre le cœur gauche et le cœur droit, il est également influencé par le terrain :
- La constitution d'une hypertension artérielle pulmonaire fixée est d'autant plus précoce que le siège de la communication anormale est proche des poumons. Ainsi, le risque est-il important et précoce (quelques mois) en présence d'un canal artériel, plus tardif (classiquement pas avant un an) en cas de communication inter-ventriculaire et inconstant et très tardif (à l'âge adulte) en présence d'une communication inter-auriculaire. Ceci fait soupçonner un rôle toxique surajouté de l'oxygène contenue dans le sang abordant la circulation pulmonaire (où le sang est habituellement pauvre en O2).
- En fonction du terrain, cette évolution peut survenir plus tôt, en particulier chez les enfants porteurs d'une trisomie 21 (syndrome de Down). Exceptionnellement, elle peut s'installer dès la naissance par « persistance de résistances pulmonaires de type fœtal ».
Symptômes
Il faut comprendre qu'initialement au moins, la constitution d'une hypertension artérielle pulmonaire fixée peut être considérée comme un mécanisme de défense que l'organisme oppose à l'hyperdébit pulmonaire responsable d'œdème pulmonaire et d'insuffisance cardiaque. Historiquement, avant la chirurgie, c'était grâce à ce mécanisme que survivaient les enfants présentant un shunt gauche-droit important. L'élévation des pressions pulmonaires tend en effet à diminuer la différence de pression entre le cœur droit et le cœur gauche et donc l'importance du shunt gauche-droit.
Ainsi, à sa phase initiale, un syndrome d'Eisenmenger s'accompagne d'une diminution puis disparition des manifestations d'insuffisance cardiaque présentée par l'enfant (dyspnée, difficultés alimentaires…) et des anomalies d'auscultation (importance du souffle cardiaque, présence d'un bruit de galop…) qui traduisaient la présence d'un shunt gauche-droit important. Cependant, le développement de certains signes tels qu'un éclat du deuxième bruit cardiaque au foyer pulmonaire (traduisant la présence d'une hypertension pulmonaire) ou d'une hypertrophie ventriculaire droite sur l'électrocardiogramme montre qu'il s'agit d'une « fausse » guérison.
Le syndrome associe :
- une cyanose (extrémités bleutées), d'abord intermittente, liée à l'effort puis constante, mais plus ou moins marquée en fonction du degré de la polyglobulie (augmentation du nombre de globules rouges. Il peut exister un hippocratisme digital ;
- une dyspnée (difficulté à respirer) et asthénie (fatigue), liées à l'hypoxie (manque d'oxygène) des tissus de l'organisme, en particulier des muscles et donc limitant les possibilités d'effort ;
- des céphalées (maux de tête), liées elles aussi à l'hypoxie tissulaire, mais également à l'augmentation de la viscosité sanguine secondaire à la polyglobulie.
Signes cliniques et paracliniques
Initialement, l'examen clinique montre une disparition progressive des signes d'insuffisance cardiaque secondaires au shunt gauche-droit. Pendant une longue phase, souvent plusieurs dizaines d'années, le patient ne présente aucune manifestation d'insuffisance cardiaque. Ce n'est qu'à une phase avancée qu’apparaîtront des signes d'insuffisance cardiaque droite liés à la défaillance progressive du ventricule droit.
La biologie confirme l'hypoxie, retrouve une polyglobulie avec une carence martiale[3]. La thrombopénie est de mauvais pronostic[4].
L'électrocardiogramme permet de suivre le développement de signes d'hypertrophie du ventricule droit et de l'oreillette droite parallèles à l'aggravation de l'obstacle pulmonaire. Tardivement, il peut également montrer des signes d'ischémie myocardique.
La radiographie thoracique montre, par des signes indirects, l'altération de la vascularisation pulmonaire : dilatation (parfois ectasique) des artères pulmonaires proximales, raréfaction des artères et artérioles pulmonaires distales (périphérie « claire »).
L'échocardiographie permet d'apprécier l'importance et de suivre l'évolution de l'hypertension pulmonaire par des estimations indirectes des pressions artérielles pulmonaires systolique (par mesure Doppler de la vitesse du flux au travers d'une insuffisance de la valve tricuspide) et diastolique (par la même mesure au travers de l'insuffisance des valves pulmonaires). Il permet également d'apprécier l'adaptation du ventricule droit à la surcharge de pression (épaisseur et cinétique des parois, diamètre de la cavité) et de dépister une défaillance de celui-ci (apparition d'une insuffisance tricuspide anormale, signes de stase veineuse).
Le cathétérisme cardiaque permet de mesurer directement le niveau des pressions et des résistances pulmonaires et de montrer leur insensibilité à diverses substances vasodilatatrices (en particulier le monoxyde d'azote), confirmant ainsi le caractère « fixé » de l'hypertension pulmonaire. L'opacification des artères pulmonaires par produit de contraste (artériographie pulmonaire) afin de juger de leurs altérations, est le plus souvent déconseillée car source potentielle d'accident. Le cathétérisme d'une veine pulmonaire (qui nécessite la présence d'un foramen ovale perméable pour que la sonde puisse passer dans l'oreillette gauche puis dans une des veines pulmonaires qui s'y abouchent) permet de s'assurer du caractère précapillaire de l'hypertension artérielle pulmonaire : la pression régnant dans les capillaires et les veines pulmonaires est alors normale ou basse. L'injection sous pression contrôlée de produit de contraste dans cette veine pulmonaire permet d'opacifier de façon rétrograde les artères pulmonaires distales et de montrer un aspect « en arbre mort » typique.
Dans les cas difficiles, c'est-à-dire quand ces explorations n'ont pas suffi à démonter le caractère fixé et donc irréversible de l'hypertension artérielle pulmonaire, on peut recourir à l'analyse microscopique d'un fragment de poumon prélevé par biopsie pulmonaire, faite habituellement de manière chirurgicale.
Complications
Le pronostic est sensiblement meilleur que celui des hypertensions artérielles pulmonaires primitives, avec des survies pouvant atteindre plusieurs décennies[5]. La mortalité atteint toutefois 70% sur 10 ans[6].
Complications pulmonaires
Les artères pulmonaires fragilisées peuvent être le siège de rupture ou de thrombose.
- La rupture se manifeste par une hémoptysie (toux avec expectoration de sang rouge) plus ou moins abondante selon son siège. Classiquement, l'apparition d'hémoptysies était un signe péjoratif à relativement court terme. En fait, il peut s'agir d'accidents ponctuels et transitoires, mais sans suite.
- La thrombose ou plutôt les thromboses (occlusion par un caillot) surviennent préférentiellement sur les artères distales et sont le plus souvent méconnues. Amputant le champ pulmonaire, elles majorent les troubles de l'hématose (échanges gazeux entre le sang et les espaces aériens pulmonaires) et donc l'hypoxie tissulaire. Elles concourent également à une majoration supplémentaire de l'hypertension artérielle pulmonaire. Elles surviennent dans environ un cas sur cinq, et d'autant plus que le patient est âgé ou qu'il existe une défaillance des deux ventricules[7].
Complications cardiaques
C'est essentiellement la défaillance du ventricule droit et l'apparition d'une insuffisance cardiaque droite. Celle-ci est habituellement beaucoup plus tardive qu'en présence d'une hypertension artérielle pulmonaire primitive et ne survient qu'après plusieurs décennies.
Il faut également mentionner la possibilité de survenue d'un angor ou de troubles du rythme cardiaque (atrial ou ventriculaire[8]) pouvant occasionner une mort subite[9], essentiellement à l'effort, traduisant une ischémie (manque d'oxygène) myocardique secondaire à une insuffisance coronarienne fonctionnelle, c'est-à-dire, sans lésion des artères coronaires.
Complications générales
La polyglobulie (augmentation du nombre de globules rouges) correspond à une réaction de l'organisme à l'hypoxie dans une tentative d'améliorer celle-ci par une augmentation des transporteurs d'oxygène (l'hémoglobine contenue dans les globules rouges). D'intensité variable, elle a tendance à provoquer ou aggraver les manifestations suivantes : cyanose, céphalées, asthénie, vertiges voire syncope liés à l'hyperviscosité sanguine. Elle peut se compliquer d'une thrombose, artérielle pulmonaire ou veineuse (et comportant alors le risque d'embolie pulmonaire) ou de saignements, ce qui peut paraître paradoxal, mais la polyglobulie secondaire aux cardiopathies congénitales cyanogènes s'accompagne souvent d'anomalie de la fonction ou du nombre des plaquettes sanguines.
Des embolies paradoxales peuvent survenir : normalement, la migration après détachement d'un thrombus formé dans une veine ne peut se faire que dans le circuit veineux, le cœur droit puis la voie pulmonaire. En présence d'un shunt droit-gauche, ce même thrombus peut gagner la grande circulation au travers de la communication et se bloquer dans un organe tel que le cerveau, la rate, un rein, etc. Le terme paradoxal tient au point de départ veineux et au point d'arrivée artériel. Le thrombus en cause peut être cruorique (caillot sanguin) et à l'origine d'un accident vasculaire cérébral, d'un infarctus splénique, etc. Il peut être également constitué de matériel infectieux et alors à l'origine d'abcès infectieux, tout particulièrement cérébraux.
Des hémorragies sont fréquentes, habituellement peu graves[3].
La grossesse
La présence d'un syndrome d'Eisenmenger est une des très rares cardiopathies congénitales qui contre-indique formellement la grossesse.
En effet, la grossesse précipite l'évolution naturelle de la maladie[10] ; c'est-à-dire qu'au mieux elle aggraverait l'état de la mère, au pire elle pourrait être responsable de son décès[11] (non exceptionnel). En fonction de la gravité de l'hypoxie et de la cyanose, Il y a également un risque important que la grossesse n'arrive pas à son terme (mort fœtale in utero) ou fasse naître un enfant très dysmature.
L'évolution naturelle d'une hypertension artérielle pulmonaire fixée secondaire à une cardiopathie, habituellement étalée sur plusieurs décennies, autorise par contre le plus souvent une procédure d'adoption.
Traitement
La prise en charge du syndrome d'Eisenmenger a fait l'objet de la publication de recommandations dans la cadre des cardiopathies congénitales de l'adulte. Celles, européennes, datent de 2020[12], celles, américaines, de 2018[13].
Traitements non spécifiques
La prévention de l'endocardite bactérienne, recommandée pour toutes les cardiopathies congénitales est ici d'autant plus impérieuse que coexiste un risque d'embolie paradoxale et que tous les organes peuvent être l'objet d'une colonisation bactérienne.
La prévention de la thrombose fait appel soit au traitement anti-coagulant par voie orale (Anti-vitamines K), soit à un traitement anti-agrégeant plaquettaire par Aspirine. Le choix et les modalités pratiques du traitement sont rendus délicats par la majoration du risque d'hémorragies intra-pulmonaires et les troubles de la fonction plaquettaire fréquemment associés[3]. L'utilisation d'un anticoagulant oral direct pourrait augmenter le risque de survenue d'une complication cardiaque et n'est pas recommandée[14].
Au cours d'une polyglobulie, il est exceptionnel que l'on ait recours aux traitements usités dans la maladie de Vaquez (ou polyglobulie essentielle) en raison du risque d'atteinte des autres lignées sanguines (globules blancs et plaquettes) qu'ils entrainent. Seule se discute la saignée. Celle-ci est controversée car elle est à l'origine d'un déficit chronique en fer et n'a qu'un effet transitoire car elle stimule en réaction l’érythropoïèse (fabrication de globules rouges par l'organisme). Actuellement, la pratique d'une saignée est moins décidée sur des critères biologiques (nombre de globules rouges, taux d'hémoglobine, hématocrite) que sur l'importance des manifestations cliniques d'hyperviscosité sanguine : céphalées, asthénie, vertiges, etc. Quand elle est retenue, on procède à la soustraction lente de 300 à 400 cm3 de sang avec compensation par la même quantité d'un soluté de remplissage.
Un apport en fer est recommandé en cas de microcytose (globules rouges plus petits que normalement) car une polyglobulie microcytaire expose plus à la thrombose qu'une polyglobulie équivalente faite de globules rouges normocytaires.
La dégradation exagérée des globules rouges peut être à l'origine d'une hyperuricémie (taux anormalement élevé d'acide urique dans le sang) et éventuellement de crise de goutte ou de lithiase rénale. Cette hyperuricémie peut être combattue par divers traitements, en particulier l'Allopurinol qui agit en réduisant la synthèse des acides nucléiques.
Il faut veiller à une hydratation correcte : en particulier par fortes chaleurs, les patients présentant un syndrome d'Eisenmenger devront boire abondamment dans le double but d'éviter une majoration de l'hyperviscosité sanguine par hémoconcentration et de favoriser l'élimination rénale de l'acide urique en excès.
Le traitement de l'insuffisance cardiaque repose sur les diurétiques et, éventuellement, les digitaliques[15].
Un exercice physique important n'est pas recommandé[15]. Une réadaptation à l'effort serait cependant utile[3].
Traitements de l'hypertension pulmonaire
Il repose sur l'utilisation de médicaments vaso-dilatateurs ayant un effet sur la circulation pulmonaire. Ceux-ci sont administrés soit par voie orale, soit en perfusion intra-veineuse continue. Ces traitements ont fait la preuve de leur efficacité dans l'hypertension artérielle pulmonaire primitive ou secondaire à une maladie de système mais n'ont pas été étudiés aussi spécifiquement dans le cadre du syndrome d'Eisenmenger. Ils peuvent être donnés seuls ou en combinaison.
- le bosentan ou Tracleer, antagoniste des récepteurs A et B de l'endothéline I, administré en 2 prises orales quotidiennes[16]. Il est donné en première intention[3].
- l'époprosténol ou Flolan, prostacycline synthétique analogue de la Prostaglandine I2, administré en perfusion intra-veineuse continue à l'aide de mini-pompes portables,
- l'iloprost, autre prostanoide, administré sous forme inhalée 6 à 9 fois par jour,
- le sildenafil ou Revatio, inhibiteur de la phosphodiestérase type 5 (équivalent du Viagra), administré en 3-4 prises orales quotidiennes[17].
Il s'agit de traitements particulièrement onéreux, contraignants pour les patients, non dénués d'effets secondaires et nécessitant de strictes précautions d'emploi. Leur prescription relève de médecins spécialisés dans la prise en charge de l'hypertension artérielle pulmonaire. Ils permettent toutefois de retarder l'évolution vers la greffe[18] et ont une certaine efficacité sur les symptômes des formes évoluées[19].
Traitement chirurgical
Il s'agit de la greffe cœur-poumons, qui ne sera envisagée qu'en dernier ressort après avoir épuisé les diverses possibilités médicales, d'autant plus que les diverses interventions que le patient aura eu dans son enfance sur sa cardiopathie peuvent en rendre la réalisation plus difficile et le résultat plus aléatoire. Une solution alternative est la transplantation pulmonaire avec fermeture chirurgicale du shunt responsable, avec des résultats comparables avec la greffe coeur-poumons et une survie pouvant dépasser 15 ans[20]. Cela est surtout vrai en cas de communication interatriale où les chances de récupération d'un fonction ventriculaire droite correcte sont plus importantes[21].
Notes et références
- Eisenmenger V, Die angeborenen Defecte der Kammerscheidewand des Herzens, Z Klin Med, 1897;32:1-28
- (en) Pietra GG, Capron F, Stewart S et al. Pathologic assessment of vasculopathies in pulmonary hypertension, J Am Coll Cardiol, 2004;43(Suppl S):25S–32S
- Arvanitaki A, Gatzoulis MA, Opotowsky AR et al. Eisenmenger Syndrome, J Am Coll Cardiol, 2022;79:1183-1198
- Martin-Garcia AC, Arachchillage DR, Kempny A et al. Platelet count and mean platelet volume predict outcome in adults with Eisenmenger syndrome, Heart, 2018,104:45-50
- (en) Hopkins WE, Ochoa LL, Richardson GW, Trulock EP, Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome, J Heart Lung Transplant, 1996;15:100–105
- Diller GP, Körten MA, Bauer UM et al. Current therapy and outcome of Eisenmenger syndrome: data of the German National Register for congenital heart defects, Eur Heart J, 2016;37:1449-1455
- (en) Broberg CS, Ujita M, Prasad S et al. Pulmonary arterial thrombosis in Eisenmenger syndrome is associated with biventricular dysfunction and decreased pulmonary flow velocity, J Am Coll Cardiol, 2007;50:634-642
- Baskar S, Horne P, Fitzsimmons S et al. Arrhythmia burden and related outcomes in Eisenmenger syndrome, Congenit Heart Dis, 2017;12:512-519
- Daliento L, Somerville J, Presbitero P et al. Eisenmenger syndrome. Factors relating to deterioration and death, Eur Heart J, 1998;19:1845-1855
- D'Alto M, Diller GP, Pulmonary hypertension in adults with congenital heart disease and Eisenmenger syndrome: current advanced management strategies, Heart, 2014;100:1322–1328
- Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J et al. 2018 ESC guidelines for the management of cardiovascular diseases during pregnancy, Eur Heart J, 2019;39:3165-3241
- Baumgartner H, De Backer J, Babu-Narayan SV et al. 2020 ESC guidelines for the management of adult congenital heart disease, Eur Heart J, 2021;4:563-645
- Stout KK, Daniels CJ, Aboulhosn JA et al. 2018 AHA/ACC guideline for the management of adults with congenital heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines, J Am Coll Cardiol, 2019;73:1494-1563
- Freisinger E, Gerss J, Makowski L et al. Current use and safety of novel oral anticoagulants in adults with congenital heart disease: results of a nationwide analysis including more than 44 000 patients, Eur Heart J, 2020;41:4168-4177,
- (en) Beghetti M, Galiè N, Eisenmenger syndrome: A clinical perspective in a new therapeutic era of pulmonary arterial hypertension, J Am Coll Cardiol, 2009;53:733-740
- (en) Galiè N, Beghetti M, Gatzoulis MA et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study, Circulation, 2006;114:48–54
- (en) Chau EM, Fan KY, Chow WH, Effects of chronic sildenafil in patients with Eisenmenger syndrome versus idiopathic pulmonary arterial hypertension, Int J Cardiol, 2007;120:301-305
- (en) Adriaenssens T, Delcroix M, Van Deyk K, Budts W, Advanced therapy may delay the need for transplantation in patients with the Eisenmenger syndrome, Eur Heart J, 2006;27:1472-1477
- Opotowsky AR, Landzberg MJ, Beghetti M, The exceptional and far-flung manifestations of heart failure in Eisenmenger syndrome, Heart Fail Clin, 2014;10:91–104
- Hjortshoj CS, Gilljam T, Dellgren G et al. Outcome after heart-lung or lung transplantation in patients with Eisenmenger syndrome, Heart, 2020;106:127-132
- Sertic F, Han J, Diagne D et al. Not all septal defects are equal: outcomes of bilateral lung transplant with cardiac defect repair vs combined heart-lung transplant in patients with Eisenmenger syndrome in the United States, Chest, 2020;158:2097-2106