Principe d'équivalence temps-température
Le principe d’équivalence temps-température permet de préciser la dépendance à la température des fonctions (ou grandeurs) viscoélastiques linéaires telles E(t), E'(f), E''(f) et |η*|(f).
Dans le cas d’essais périodiques, l’effet de la fréquence de sollicitation (ou de la vitesse de sollicitation dans le cas par exemple d’un essai de traction uniaxiale) sur les grandeurs viscoélastiques d’un polymère amorphe est inverse à celui de la température. Les effets de la fréquence et de la température sont donc dépendants. Ce phénomène est à l’origine du principe d’équivalence temps-température en viscoélasticité linéaire.
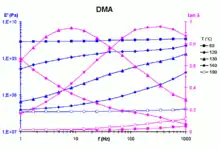
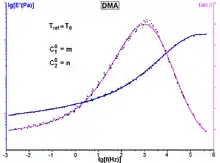
Après avoir déterminé des courbes isothermes grandeurs viscoélastiques-fréquence et ce à plusieurs températures (par exemple en analyse mécanique dynamique, voir Fig. 1), ce principe peut être utilisé, en faisant intervenir un facteur de translation, pour corréler ces grandeurs dans toute la gamme de température et de fréquence de mesure. L’application de ce principe permet aussi le calcul, à partir des données expérimentales, de courbes maîtresses montrant l’effet de la fréquence sur un très large intervalle[1].
Ce principe et la loi établie par Malcolm L. Williams, Robert F. Landel et John D. Ferry (ou loi de WLF) permettent de déterminer les coefficients intrinsèques C1 et C2 d’un polymère.
La loi WLF et une loi d'Arrhenius donnent une équivalence entre le temps et la température. La première est liée à des mouvements moléculaires globaux, la seconde à des mouvements plus locaux. Une conséquence pratique de cette mobilité moléculaire est la durée limitée de conservation des aliments[1].
Principe physique
Un polymère (corps viscoélastique) est soumis à une sollicitation dynamique. Si la fréquence d’excitation est suffisamment basse (temps très supérieur au temps caractéristique τ appelé aussi temps de relaxation, fonction de la masse moléculaire), le comportement visqueux est prépondérant et toutes les chaînes ont le temps de « répondre » au cours d’une période de contrainte. Par contre, à plus haute fréquence, les chaînes n’ont pas le temps de répondre totalement et le blocage artificiel qui en résulte se traduit par une augmentation du module.
D’autre part, à fréquence constante, une augmentation de la température se traduit par une diminution du module, à la suite de l’augmentation des volumes libres et des mouvements de chaînes.
Les composantes M' et M'' d’un module complexe M* (tel E* ou G*) expérimental dépendent notamment de la fréquence et de la température.
Principe d'équivalence temps-température
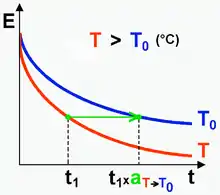
On considère le module de relaxation E à deux températures T et T0 telles que T > T0. À déformation constante, la contrainte relaxe plus vite à la température la plus élevée (Fig. 2).
Le principe d’équivalence temps-température stipule que le passage de T à T0 (soit un changement de température) revient à multiplier l’échelle du temps par un facteur constant uniquement fonction des deux températures T et T0[3]


où P est la propriété considérée.
Ce qui peut se formuler par l’égalité du module pour des couples temps/températures différents[4]

(noté aussi aT) est appelé facteur de translation (horizontale)[5] (shift factor en anglais).
Si T > T0 aT > 1 (Fig. 2),

si T < T0 aT < 1,
si T = T0 aT = 1 (Fig. 3).
La forme du principe d’équivalence (t, T) pour les modules dynamiques s’obtient de manière similaire


Une diminution de température augmente les temps caractéristiques et diminue les fréquences caractéristiques.
Relation entre le facteur de translation et les viscosités limites
Pour un polymère en solution ou à l’état « fondu », on montre la relation suivante entre les viscosités en écoulement continu (sur le premier plateau newtonien)[6]


où est la viscosité en écoulement continu à la température T.

Facteur de translation utilisant la loi WLF
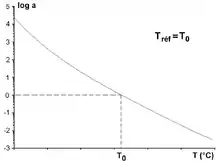
La relation empirique de Williams-Landel-Ferry (relation ou loi WLF, publiée en 1955), associée au principe d’équivalence temps-température, permet de rendre compte des variations de la viscosité limite de polymères amorphes (non cristallins) en fonction de la température, pour des températures proches de la température de transition vitreuse Tv. La loi WLF exprime également la variation avec la température du facteur de translation.

Un traitement mathématique calcule aT pour chacune des composantes M' et M'' du module complexe mesuré M*. Une bonne corrélation entre les deux facteurs de translation donne les valeurs des coefficients C1 et C2 caractéristiques de la matière. La loi WLF n’est vérifiée que dans la plage approximative de température [Tv, Tv + 100 °C]. Williams, Landel et Ferry ont proposé une équation de variation de aT en fonction de (T-T0)[3]

Les coefficients (ou constantes) et (positifs) dépendent du polymère considéré et de T0, une température de référence convenablement choisie.


Si Tréf = Tv

où (noté aussi C1v) est le coefficient C1 à Tv.

Transformation des coefficients C1 et C2 en fonction de la température :
- si passage de T0 à T'0

- en particulier, si passage de Tv à T0

et sont les coefficients de la loi WLF.

Ces mêmes auteurs ont proposé les « constantes universelles » et (pour un système polymère donné) rassemblées dans un tableau[8]. Elles sont approximativement identiques pour un grand nombre de polymères et on peut écrire 15 et 50 K. On observe expérimentalement des écarts aux valeurs du tableau[3]. Ces ordres de grandeurs (utiles) sont à respecter : ils sont un bon indicateur de la qualité d’une corrélation. Les grandeurs et doivent donc être considérées comme des paramètres ajustables[1].


Application : construction de courbes maîtresses
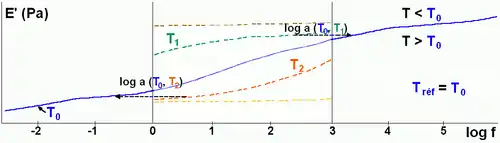
Le principe de superposition temps-température permet, dans l’hypothèse de simplicité thermorhéologique (tous les temps caractéristiques présentent la même loi de variation avec la température)[1], à partir d’une fenêtre spectrale initiale [f1, f2] et d’une série d’isothermes dans cette fenêtre, un calcul des courbes maîtresses d’un matériau qui s’étendent sur une plage de fréquence plus large. Une température arbitraire, T0, est prise comme référence pour fixer l’échelle des fréquences (la courbe à cette température ne subit aucune translation).
Dans la fenêtre [f1, f2], si la température augmente par rapport à T0, E'(f) diminue (Fig. 4), cela revient à explorer une partie de la courbe maîtresse correspondant à des fréquences inférieures à f1, en restant à T0. Inversement, baisser la température revient à explorer une partie de la courbe correspondant à des fréquences élevées.
En choisissant une température de référence (T0), les translations (horizontales) de T à T0 ont pour amplitude lg aT suivant l’axe des fréquences (Fig. 4).
Dans la zone de transition vitreuse, aT est décrit par une fonction homographique de la température.
Le comportement viscoélastique est ainsi modélisé et permet une extrapolation au-delà du domaine de fréquences expérimentales qui s’étend typiquement de 0,01 à 100 Hz.
Facteur de translation utilisant une loi d'Arrhenius
Le facteur de translation (qui dépend de la nature de la transition) peut être défini, en dessous de Tv, en utilisant une loi d'Arrhenius[10]

avec :
- Ea, l’énergie d'activation apparente ;
- R, la constante universelle des gaz parfaits ;
- et T0, une température de référence en kelvins.
Cette loi d’Arrhenius, dans cette plage sous-vitreuse, s’applique à des transitions secondaires (relaxations) dites « β ».
En utilisant les mêmes axes que ceux de la Fig. 3, une courbe de la variation de aT en fonction de T peut être tracée (à une température de référence).
Limitations
L’échantillon homogène, isotrope et amorphe doit être sollicité dans sa zone de comportement linéaire (dans ce cas, la déformation s’exprime comme une fonction linéaire de la contrainte), en lui appliquant un très faible taux de déformation, par exemple 0,01 %.
Pour vérifier la loi WLF, un tel échantillon doit être sollicité dans la plage de température approximative [Tv, Tv + 100 °C], où sont observées des transitions (relaxations) « α ». L’étude permettant de déterminer aT ainsi que les coefficients C1 et C2 nécessite de nombreux essais [en dynamique, un double balayage fréquence/température (Fig. 1), ce qui représente au minimum une centaine de points de mesure].
Notes et références
- Jo Perez, Matériaux non cristallins et science du désordre, Lausanne, PPUR, , 557 p. (ISBN 2-88074-485-7, lire en ligne), p. 143, 274, 276
- ẟ est l’angle de phase.
- (en) Jacques Verdu, Bruno Fayolle et Yves Mouton (dir.), Organic materials for sustainable construction, Londres, John Wiley & Sons, , 682 p. (ISBN 978-1-84821-224-4, lire en ligne), partie 2, chap. 5 (« Organic polymers »), p. 102
- Si l’on ne tient pas compte de la dilatation volumique.
- Synonymes : facteur (ou coefficient) de glissement, de décalage, de déplacement, de thermodépendance.
- Voir aussi l’article (de) Équation de Vogel-Fulcher-Tammann.
- Courbe générée par logiciel à partir des données d’un essai dynamique comportant un double balayage fréquence/température sur une grandeur viscoélastique d’un polymère.
- Tableau intitulé Parameters characterizing temperature dependence of aT for various polymer systems, qui donne, pour Tréf = Tv, les valeurs et de plusieurs dizaines de polymères (certains sont en solution).
- Une courbe maîtresse peut être représentée par un polynôme d’interpolation de degré 7.
- Parviz Navi et Frédéric Heger, Comportement thermo-hydromécanique du bois : Applications technologiques et dans les structures, Lausanne/Paris, PPUR, , 298 p. (ISBN 2-88074-620-5, lire en ligne), p. 116, 117
Voir aussi
Bibliographie
- (en) John D. Ferry (en), Viscoelastic properties of polymers, New York, John Wiley & Sons, , 3e éd., 641 p. (ISBN 978-0-471-04894-7, OCLC 5942663, lire en ligne)
- Jo Perez (préf. Lucien Monnerie), Physique et mécanique des polymères amorphes, Paris, Tec & Doc, , 384 p. (ISBN 978-2-85206-787-5, BNF 35504448)
- Michel Fontanille et Yves Gnanou, Chimie et physico-chimie des polymères, Paris, Dunod, coll. « Sciences Sup », , 3e éd., 576 p. (ISBN 978-2-10-058915-9)
Articles connexes
- Principe de superposition de Boltzmann
- Analyse mécanique dynamique, qui permet de générer des courbes maîtresses.
- Modèle de Kelvin-Voigt, une des modélisations les plus élémentaires d’un corps viscoélastique, faisant intervenir un temps caractéristique.
- Nombre de Deborah
- Indicateur temps-température