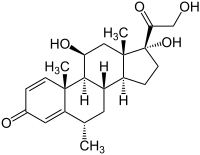
Méthylprednisolone
La méthylprednisolone (DCI), aussi appelée 6-alpha-méthylprednisolone, est un corticoïde de la famille des glucocorticoïdes (comme la prednisolone ou le cortisol). Elle est utilisée dans les traitements anti-inflammatoires (allergies…), dans les dérèglements du sang et les vomissements tardifs induits par chimiothérapie. La méthylprednisolone est la substance active du Médrol, du Solumédrol.
| Méthylprednisolone | ||
| ||
 | ||
| Identification | ||
|---|---|---|
| No CAS | (hémisuccinate) |
|
| No ECHA | 100.001.343 | |
| No CE | 201-476-4 | |
| Code ATC | D07, D10, H02 | |
| SMILES | ||
| InChI | ||
| Propriétés chimiques | ||
| Formule | C22H30O5 [Isomères] |
|
| Masse molaire[1] | 374,470 6 ± 0,021 2 g/mol C 70,56 %, H 8,07 %, O 21,36 %, |
|
| Propriétés physiques | ||
| T° fusion | 232,5 °C | |
| Unités du SI et CNTP, sauf indication contraire. | ||
Il fait partie de la liste des médicaments essentiels de l'Organisation mondiale de la santé (liste mise à jour en )[2].

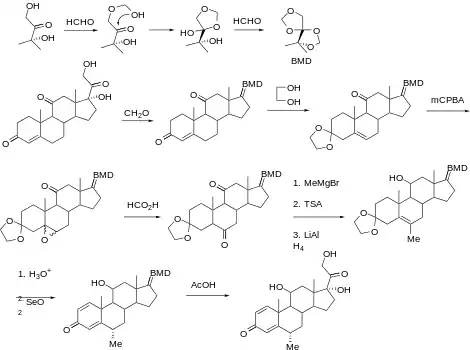
Synthèse
Pharmacocinétique
L'absorption de la méthylprednisolone
L’absorption digestive (dans la partie initiale du jéjunum) de la méthylprednisone est rapide, d’environ 80 % par voie orale après dose unique.
Fixation protéique
Dans le plasma, la méthylprednisolone circule en majorité sous forme liée (77 %) à deux protéines de transport : l’albumine, possédant une forte capacité mais une faible affinité, et la transcortine ou « Cortisol Binding Globulin » (CBG), alpha 2 globuline possédant une faible capacité et une forte affinité.
Métabolisme d'élimination
La voie métabolique d'élimination de la méthylprednisolone est mal connue. Les principales enzymes impliquées dans l’élimination hépatique de la méthylprednisolone semblent être la 11β-hydroxystéroïde deshydrogénase ainsi que la 20 céto-stéroïde réductase. La 6β -hydroxylation des corticostéroïdes est probablement une voie quantitativement mineure dans ce métabolisme. Cependant, étant dépendante du cytochrome p. 450 3A4 (CYP3A4), cette voie peut être significativement influencée par l’administration d’inducteur ou d’inhibiteur enzymatiques. Le métabolisme de la méthylprednisolone est le plus sensible aux inducteurs ou aux inhibiteurs du CYP3A4 comparé a ceux des différents autres glucocorticoïdes. Ce qui induit une série d'interactions médicamenteuses plus présentes dans le cas de la méthylprednisolone que pour les autres glucocorticoïdes.
La cinétique de la méthylprednisolone est linéaire, contrairement à la prednisone qui n'est pas dose-dépendante car elle doit subir une 11β -hydroxylation pour être transformée en prednisolone.
La demi-vie d’élimination plasmatique de la méthylprednisolone est de l’ordre de 180 minutes.
Pharmacodynamique
Relation structure-activité
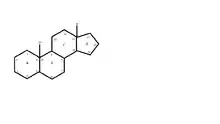
Les corticostéroïdes naturels synthétisés par les surrénales ont soit une activité glucocorticoïde, soit une activité minéralocorticoïde, soit les deux.
Les glucocorticoïdes présentent une homogénéité de structure, le noyau prégnane.
Les fonctions nécessaires à l'activité glucocorticoïdes sont les suivantes :
- cétone (C=O) en 3 ;
- cétone en 20 ;
- double liaison 4-5 sur le cycle A ;
- groupement hydroxyle (OH) en 11.
Dans le cas de la méthylprednisolone, il y a ajout d'un méthyl en alpha pour augmenter l'activité anti-inflammatoire de la molécule.
| Activité anti-inflammatoire | Activité minéralo-corticoïde | Demi-vie biologique (heure) | |
|---|---|---|---|
| Hydrocortisone | 1 | 1 | 8-12 |
| Cortisone | 0,8 | 0,8 | 8-12 |
| Prednisolone | 4 | 0,8 | 12-36 |
| Méthylprednisolone | 5 | 0,5 | 12-36 |
Propriétés anti-inflammatoire et immunosuppressive
L'action de la méthylprednisolone sur les différents acteurs de l'inflammation et de l'immunité est résumée ci-dessous :
- inhibition de la transcription des cytokines pro-inflammatoires ;
- diminution de l’acide arachidonique par la synthèse de lipocortine-1 qui possède une activité anti-phospholipase A2 ;
- inhibition de l'expression des molécules d'adhésion ;
- globules blancs :
- macrophages : diminution de leur différenciation et de leurs activités anti-infectieuses,
- polynucléaires neutrophiles : augmentation des PNN (polynucléaires neutrophiles) circulants, inhibition de l’adhésion,
- lymphocytes : diminution du taux de lymphocytes circulants ;
- diminution de la perméabilité vasculaire et de l’activation des cellules endothéliales, inhibition de l’afflux des leucocytes ;
- fibroblastes : diminution de la prolifération, diminution de la production de protéines (collagène).
Contrôles apportés au principe actif pour une commercialisation
De nombreux contrôles sont apportés aux principes actifs et notamment à la méthylprednisolone. Parmi les contrôles effectués, en voici quelques-uns :
- température de fusion (environ 223,5 °C) ;
- spectrophotométrie (UV, IR) ;
- solubilité dans différents solvants (eau, alcool, dioxane, méthanol, acétone, chloroforme, éther) ;
- rotation spécifique (comprise entre +79° et +86°) ;
- résistance à la dessiccation (<1,0 % de pertes) ;
- résidu à l'allumage (<0,2 % de résidus) ;
- pureté chromatographique (<1,0 % impureté individuelle ; <2,0 % impuretés totales) ;
- analyse quantitative (97 % à 103 %).
Ces contrôles sont effectués chez le fabricant Pfizer avant commercialisation.
Mode d'action
Effet génomique
Les glucocorticoïdes se retrouvent dans la circulation sanguine soit sous forme libre, soit liés à une protéine plasmatique. Environ 80 % de la méthylprednisolone est sous forme liée dans la circulation sanguine. Elle se fixe sur deux types de protéines de transport : l'albumine, possédant une forte capacité mais une faible affinité, ou la transcortine ou « Cortisol Binding Globulin » (CBG), possédant une faible capacité mais une forte affinité. Seule la forme libre de méthylprednisolone peut traverser la membrane par diffusion simple pour ensuite aller se fixer sur son récepteur à glucocorticoïde (GR).

Le récepteur au glucocorticoïdes est intracellulaire et est fixé à la protéine HSP 90 et immunophiline. Il possède 3 domaines : un domaine pour la fixation du ligand (partie C-terminal), un autre domaine pour la liaison à l'ADN (partie intermédiaire) et un dernier domaine pour l'activation du gène/régulation transcriptionnelle (N-terminal). Le gène codant le GR, composé de neuf exons, est localisé sur le chromosome 5. Un épissage alternatif dans l’exon 9 génère deux isoformes du récepteur, hautement homologues, nommées α et β. Elles sont identiques jusqu’à l’aminoacide 727 puis elles divergent : l’isoforme α (GRα) comportant 50 aminoacides supplémentaires (formant une protéine de 777 aminoacides) contre seulement 15 aminoacides non homologues (formant une protéine de 742 aminoacides) pour l’isoforme β (GRβ). L’isoforme α correspond au GR classique fonctionnant comme facteur de transcription ligand-dépendant alors que l’isoforme β ne fixe pas les agonistes glucocorticoïdes et exerce un effet dominant négatif sur l’activité transcriptionnelle du GRα. Le GRβ est impliqué dans le développement de l’insensibilité aux glucocorticoïdes dans diverses pathologies, associées pour la plupart à une dysrégulation de la fonction immunitaire.
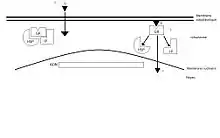
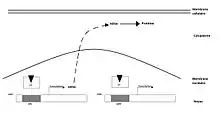
Le glucocorticoïde traverse la membrane cellulaire par diffusion simple. La fixation de l’agoniste va conduire à la dissociation du complexe GR-HSP 90-immunophiline permettant son transfert nucléaire. C’est au sein de ce noyau que le complexe hormone/récepteur va se fixer, au moyen de deux structures dites en « doigts de zinc » (portions très conservées entre tous les récepteurs des hormones stéroïdes), sur les éléments accepteurs du génome. Le complexe L-R interagit avec l'ADN sur le site « Glucocorticoids-Responsive-Elements » ou GRE. Cela stimule la transcription de lipocortine. Celle-ci inhibe la phospholipase A2, une enzyme (une hydrolase) qui libère spécifiquement l'acide gras qui estérifie la fonction alcool secondaire du glycérol d'un phosphoglycéride. Si cet acide gras est l'acide arachidonique, sa métabolisation dans l'organisme peut aboutir aux prostaglandines et aux leucotriènes, médiateurs de l'inflammation.
En produisant la lipocortine-1 qui inhibe la phospholipase A2, on comprend alors comment les glucocorticoides contribuent à faire diminuer l'inflammation. Ce sont des anti-inflammatoires stéroïdiens (AIS).
Mais elle induit aussi la répression de gènes tels ceux qui codent l’ACTH (phénomène à l’origine du rétrocontrôle négatif exercé par le cortisol), de nombreuses cytokines (molécules impliquées dans divers processus immunologiques) ou de collagénases et de la stromélysine (enzymes impliquées en particulier dans la destruction des cartilages dans les arthropathies inflammatoires).
Effet non génomique
Certains effets des corticoïdes pourraient être dus à des effets non génomiques sur l'AMP cyclique intracellulaire et sur les transports ioniques. Ces effets surviennent rapidement et donc ne peuvent résulter d'une synthèse de protéines qui nécessite plusieurs heures. Ils apparaissent pour des doses plus élevées (bolus). Citons par exemple l'action rapide des corticoïdes sur la crise d'asthme, la régression rapide des lésions tissulaires lors d'un traumatisme médullaire aigu. L’action non génomique des GRs est de mieux en mieux décrite, par des mécanismes directs et indirects faisant intervenir des interactions protéines–protéines, entre protéines de membrane ou intracytoplasmiques, avec des transporteurs des glucocorticoïdes. Ainsi, il existerait des interactions spécifiques des glucocorticoïdes avec des GR présents au niveau des membranes cellulaires et des effets non génomiques médiés par fixation aux GR cytosoliques. Par exemple, les glucocorticoïdes inhiberaient la recapture de la noradrénaline médiée par le transporteur extraneuronal des monoamines.
Activité anti-inflammatoire
Les principales cellules cibles sont les polynucléaires neutrophiles circulants, les macrophages et les cellules fibroblastiques locales.
Les corticoides agissent aussi sur les mécanismes de l'inflammation :
- réduction de la vasodilatation et de l'œdème, par inhibition de la production des agents vasoactifs : histamine, bradykinine, leucotriène C, prostaglandines ;
- diminution du chimiotactisme et de l'afflux des leucocytes et monocytes au site de l'inflammation ;
- diminution des fonctions des cellules phagocytaires (monocytes/macrophages), diminution de la sécrétion d'interleukine-l (IL-1), de TNF-a et d'INF-g, stabilisation des membranes lysosomiales (inhibition de la libération des enzymes protéolytiques).
Activité anti-allergique
Inhibition de la dégranulation des mastocytes et des basophiles (augmentation du taux d'AMP, cyclique intracellulaire).
Activité immunosuppressive
Inhibition des réactions d'hypersensibilité retardée mais les corticoïdes ne modifient pas la production d'anticorps.
Inhibition de la multiplication des lymphocytes, surtout lymphocytes T CD4+, notamment par inhibition de la synthèse des cytokines pro-inflammatoires activées (IL-1, IL-6, IL-2, INF et TNF).
La baisse de la synthèse d'IL-1 et d'INFg provoque une raréfaction des molécules d'histocompatibilité à la surface des cellules présentatrices d'antigène.
Action sur l'axe hypothalomo-hypophyso-surrénalien (HHS)
Les corticoïdes freinent l'axe HHS et cette action est d'autant plus forte avec les composés à demi-vie longue.
Il existe une relation directe entre l'activité anti-inflammatoire et la capacité de bloquer l'axe HHS. De même l'apport d'un radical fluor ou méthyl augmente l'activité anti-inflammatoire donc freine d'autant plus l'axe HHS.
Interaction médicamenteuse et effets secondaires
Interaction médicamenteuse
La méthylprednisolone, un substrat des isoenzymes du cytochrome p. 450 (CYP), est métabolisée essentiellement par la CYP3A4. Elle catalyse la 6β-hydroxylation des stéroïdes, réaction de phase I essentielle à la biotransformation de la méthyprednisolone. De nombreuses molécules (médicamenteuses ou non) sont des substrats de la CYP3A4 ; certaines d’entre elles agissent sur la biotransformation des glucocorticoïdes par induction (régulation positive) ou inhibition de la CYP3A4.
Inhibiteurs de la CYP3A4 – les médicaments qui inhibent l’activité de la CYP3A4 accroissent la concentration plasmatique de la méthyprednisolone. Lors de l’administration concomitante d’un inhibiteur de la CYP3A4, il convient de réduire la dose de méthylprednisolone pour éviter tout effet toxique.
Inducteurs de la CYP3A4 – les médicaments qui induisent l’activité de la CYP3A4 diminuent la concentration plasmatique de méthylprednisolone. Ainsi, lors de l’administration concomitante, il pourrait être nécessaire d’augmenter la dose de méthylprednisolone pour obtenir l’effet escompté.
Substrats de la CYP3A4 – en présence d’un autre substrat de la CYP3A4, la clairance hépatique (métabolisme hépatique) de la méthylprednisolone peut être modifiée ; la posologie doit donc être ajustée en conséquence. Il est possible que les effets indésirables de chacun des médicaments soient plus susceptibles de se manifester au cours d’une administration concomitante.
Antibiotique (Isoniazide) – inhibiteur de la CYP3A4. De plus, la méthylprednisolone peut augmenter le taux d’acétylation et la clairance de l’isoniazide. Antibiotique (Rifampine) – inducteur de la CYP3A4. Anticoagulants (oraux).
L’effet de la méthylprednisolone sur les anticoagulants oraux est variable. La réponse de la warfarine est inhibée par les glucocorticoïdes. Par conséquent, pour assurer l’effet anticoagulant désiré, il faut surveiller les indices de coagulation. Anticonvulsivants (Carbamazépine) – inducteur et substrat de la CYP3A4. Anticonvulsivants (Phénobarbital et Phénytoïne) – inducteurs de la CYP3A.
Anticholinergiques – les corticostéroïdes peuvent modifier l’effet des anticholinergiques.
- Des cas de myopathie aiguë ont été signalés lors de la prise concomitante de fortes doses de corticostéroïdes et d’anticholinergiques.
- On a noté une suppression des effets de blocage neuromusculaire du pancuronium et du vécuronium chez des patients sous corticothérapie. On peut s’attendre à une telle interaction lors de l’utilisation de tout agent de blocage neuromusculaire agissant par antagonisme compétitif.
Antiémétiques (Aprépitant et Fosaprépitant) – inhibiteurs et substrats de la CYP3A4.
Antifongiques (Itraconazole et Kétoconazole) – inhibiteurs et substrats de la CYP3A4.
Antiviraux – inhibiteurs et substrats de la CYP3A4.
Inhibiteurs de l’aromatase (Aminoglutéthimide) – l’aminoglutéthimide peut provoquer la perte de la suppression surrénale causée par la corticothérapie.
Cholestyramine – la cholestyramine peut augmenter la clairance des corticostéroïdes.
Bloqueurs des canaux calciques (Diltiazem) – inhibiteur et substrat de la CYP3A4. Contraceptifs (oraux) (Éthynilestradiol et Noréthindrone) – inhibiteur et substrat de la CYP3A4. Les estrogènes peuvent diminuer la bio-transformation hépatique de certains corticostéroïdes, et par voie de conséquence, accentuer les effets de ces derniers.
Glucosides digitaliques – les patients qui prennent un glucoside digitalique sont exposés à un risque accru d’arythmie associé à l’hypokaliémie.
Immunosuppresseurs (Cyclosporine) – inhibiteur et substrat de la CYP3A4.
- Compte tenu de l’inhibition réciproque de la bio-transformation qui s’opère lorsque la méthylprednisolone et la cyclosporine sont administrées de façon concomitante, il se peut que la concentration plasmatique de l’un ou de l’autre médicament, ou des deux, augmente. Il est par conséquent possible que les manifestations indésirables soient plus susceptibles de se produire.
- Des convulsions ont été signalées en association avec l’administration concomitante de méthylprednisolone et de cyclosporine.
Immunosuppresseurs (Cyclophosphamide et Tacrolimus) – substrats de la CYP3A4. Kétoconazole – il a été rapporté que le kétoconazole entraîne une diminution notable de la bio-transformation de certains corticostéroïdes (pouvant atteindre 60 %) ; par conséquent, le risque d’effets indésirables associés aux corticostéroïdes devient plus important.
Macrolides (Clarithromycine et érythromycine) – inhibiteurs et substrats de la CYP3A4.
Macrolides (Troléandomycine) – inhibiteur de la CYP3A4.
AINS (anti-inflammatoires non stéroïdiens) et AAS (acide acétylsalicylique) à forte dose.
- Les hémorragies et les ulcères digestifs pourraient être plus fréquents lorsqu’on administre en concomitance des corticostéroïdes et des AINS.
- La méthylprednisolone peut augmenter la clairance de l’AAS utilisé à dose élevée.
- La prise concomitante d’AAS (ou d’un autre AINS) et d’un corticostéroïde augmente le risque de manifestations indésirables gastro-intestinales. L’AAS doit être utilisé avec précaution en association avec les corticostéroïdes chez les patients souffrant d’hypoprothrombinémie.
Agents provoquant une déplétion potassique. Il faut surveiller de près les patients sous corticostéroïdes qui prennent également des agents provoquant une déplétion potassique afin de déceler l’apparition éventuelle d’une hypokaliémie. Il existe également un risque accru d’hypokaliémie lors de l’administration de corticostéroïdes en concomitance avec de l’amphotéricine B, de la xanthine ou des agonistes des récepteurs bêta2.
Vaccins – chez les patients qui suivent une corticothérapie prolongée, la réponse immunitaire aux anatoxines ainsi qu’aux vaccins vivants ou inactivés peut être moins importante en raison de l’inhibition de la réaction des anticorps. Les corticostéroïdes peuvent également potentialiser la réplication de certains micro-organismes présents dans les vaccins vivants atténués. Si possible, il est conseillé de reporter l’administration de vaccins et d’anatoxines jusqu’à ce que la corticothérapie soit terminée.
Effets secondaires
Désordres hydroélectriques :
- hypokaliémie, rétention hydrosodée.
Troubles endocriniens et métaboliques :
- anomalies du métabolisme glucidique : intolérance glucidique, manifestations d’un diabète sucré latent ;
- syndrome de Cushing ou blocage parfois irréversible de la sécrétion physiologique d’ACTH.
Troubles musculosquelettiques (liés au catabolisme protidique et aux modifications du métabolisme calcique) :
- atrophie musculaire avec faiblesse, ostéoporose, ostéonécrose aseptique de la tête du fémur, tassements vertébraux.
Troubles digestifs :
- ulcères gastroduodénaux et leur cortège de complications.
Problèmes cutanés :
- atrophie cutanée, retards de cicatrisation, purpura, acné, hypertrichose.
Anomalies neuropsychiques :
- excitation avec euphorie et troubles du sommeil (effet recherché dans le dopage par les corticoïdes), plus rarement : états dépressifs.
- phénomène de rebond suit à l'arrêt du traitement.
Appareil reproducteur :
- augmentation ou diminution du nombre de spermatozoïdes et de la motilité de ceux-ci.
Métabolisme :
- bilan azoté négatif (dû au catabolisme protéique), dyslipidémie, lipomatose, augmentation de l’appétit (pouvant entraîner un gain pondéral).
Réactions allergiques :
- hypersensibilité au médicament, réaction anaphylactique, œdème angioneurotique.
Système cardiovasculaire :
- bradycardie, arrêt cardiaque, arythmie, hypertrophie cardiaque, collapsus circulatoire.
La transactivation concerne, entre autres, les gènes dont les produits contrôlent la néoglucogenèse (glucose-6 phosphatase, phosphoénolpyruvate carboxykinase, tyrosine aminotransférase, aspartate aminotransférase), la tension artérielle et l'équilibre osmotique (angiotensinogène, récepteur à l'angiotensine, sous-unité α du canal Na+ épithélial, kinase sgk), le catabolisme protéique (glutamine synthétase, angiotensinogène) et la pression intraoculaire (protéine TIGR). L'activation de ces gènes par les GC serait impliquée dans la survenue des effets secondaires suivant : diabète sucré, hypertension artérielle, hypokaliémie, rétention hydrosodée, fonte musculaire et glaucome.
Méthylprednisolone pour les femmes enceintes
Les données chez les femmes enceintes traitées par corticoïdes, quelles que soient la molécule et la voie d’administration, sont rassurantes (les publications sont nombreuses et le recul est important). On peut considérer à ce jour que la survenue de fentes faciales avec les corticoïdes n’est pas retenue.
Des retards de croissance intra-utérins et des petits poids de naissance ont été signalés chez des enfants de mère traitée au long cours par des corticoïdes (par voie générale ou locale) dans le cadre de pathologies chroniques (lupus, asthme, greffe d’organe…). Le rôle propre de la maladie ne peut être exclu.
Il n’est pas justifié d’arrêter ou de modifier un traitement par méthylprednisolone quelle que soit sa voie d’administration en prévision d'une grossesse.
La méthylprednisolone peut-être utilisée à posologie efficace quels que soient le terme de la grossesse et la voie d’administration.
La quantité de méthylprednisolone ingérée via le lait est faible : l’enfant reçoit environ 1 % de la dose maternelle (en mg/kg).
Pour des posologies maternelles inférieures à 50 mg/j de prednisone ou prednisolone (molécules proches), les concentrations dans le lait sont le plus souvent négligeables 3 à 4 heures après la prise maternelle. Il faut donc attendre 4 heures entre la prise de méthylprednisolone et la tétée en cas de bolus ou de traitement chronique.
Cas d'usage
La méthylprednisolone est principalement utilisé pour ses effets anti-inflammatoires. Cependant, les glucocorticoïdes ont un large éventail d'effets, y compris des changements au métabolisme et aux immuno-réactions. La liste des cas d'utilisation de la méthylprednisolone à des fins médicales est plutôt longue et est semblable à d'autres corticostéroides tels que le prednisolone. Les utilisations communes incluent la thérapie d'arthrite et le traitement à court terme de l'inflammation bronchique ou de la bronchite aiguë due à diverses maladies respiratoires. Elle est employée dans le traitement des périodes aiguës et de la gestion à long terme des maladies auto-immune, spécialement lupus érythémateux disséminé. Elle est également employée comme traitement pour la sclérose en plaques.
La méthylprednisolone est également prescrite pour non penetrating des blessures de moelle épinière. Elle est également employée pour la névrite vestibulaire. Après récupération d'ovules pour un cycle de fécondation in vitro, la méthylprednisolone peut être prescrite pour empêcher le corps de rejeter les embryons transférés, jusqu'à la période de l'implantation. Elle peut également être salutaire dans le traitement des patients dans l'arrêt cardiaque.
Arrêt d'un traitement à la méthylprednisolone
Lors de l'arrêt d'un traitement au corticoïde cela doit se faire de façon progressive afin d'éviter un rebond de la maladie. Un patient suivant une corticothérapie supérieure à un mois ne doit pas arrêter le traitement brutalement sous risque d'engendrer une insuffisance surrénalienne aiguë. Ce risque d'insuffisance surrénalienne aiguë est dû au fait qu'un traitement supérieur à un mois à une posologie supérieure à 7,5 mg/j inhibe la synthèse du CRF hypothalamique et de l'ACTH hypophysaire (freination hypothalamo-hypophysaire) ce qui aboutit à la suppression de la production endogène de cortisol. Le temps de récupération pour que le l'axe hypophysosurrénalien se remette en place est d'autant plus long que le traitement a été prolongé.On peut également observer une insuffisance surrénalienne durant le traitement en cas de stress.
Le sevrage après au traitement au corticoïde doit être progressif, environ une diminution de 1 mg toutes les 2 à 4 semaines, avec une possible prise de 20 mg/j d'hydrocortisone.
Posologie et modes d'administration
Il existe de nombreux risques d'incompatibilité physique avec l'acétate de méthyle, il ne faut donc pas l'utiliser en mélange ou dans une solution diluée. La solution utilisée et injectée par voie parentérale doit examiner avant l'injection afin d'éliminer toute possibilité de présence de particules étrangère et de décoloration.
Pour un effet local
Lorsque l'on utilise du DEPO-MEDROL cela n'a pas d'effet sur l'infection à proprement parler sur l'infection, Il a juste pour effet de camoufler l'infection en diminuant l'intensité de la douleur.
Polyarthrite rhumatoïde et arthrose
La quantité de DEPO-MEDROL injecté afin de diminuer l'intensité de la douleur au niveau d'une articulation dépend de deux paramètres, la taille de l'articulation et de la gravité de l'infection. Pour le traitement d'une maladie persistante il faut pratiquer des injections à intervalles de temps réguliers en prenant en compte le degré de soulagement du patient à la suite de la première injection. Il est préférable d'éviter les injections dans une articulation instable car des injections successives peuvent la détériorer.
Afin d'éviter le risque de détérioration de l'articulation il est préférable de suivre son état à l'aide d'un suivi radiologique.
Si l'on utilise un anesthésique local avant l'injection de DEPO-MEDROL, lire attentivement la notice d'emballage de l'anesthésique et prendre toutes les précautions nécessaires.
Bursite
Commencer par la préparation d'un champ stérile au niveau de la zone d'injection et faire une papule à ce niveau avec une solution de 1 % de chlorhydrate de procaïne. Ensuite introduisez dans une bourse séreuse une aiguille de calibre 20 à 24, montée sur une seringue vide, et aspirer quelques gouttes de synovie. Laisser l'aiguille en place, enlever la seringue ayant servi à l'aspiration et la remplacer par une petite seringue contenant la dose requise de DEPO-MEDROL. Une fois l'injection terminée, retirer l’aiguille et appliquer un petit pansement stérile sur la région.
Divers : kyste synovial, tendinite, épicondylite
Lorsque l'on traite des problèmes de tendinite ou de ténosynovite, il faut tout d'abord commencer par aseptiser la région d'injection. Ensuite il est important de faire l'injection de la suspension avec précaution dans la gaine plutôt que dans le corps du tendon qui est facilement localisable lorsqu'il est étiré. Pour des traitements comme l'épicondylite il faut faire l'injection de la suspension au niveau de la zone sensible. Pour les kystes des gaines tendineuses, injecter la suspension directement dans le kyste. Dans la majorité des cas il ne faut qu'une seule injection pour faire réduite le volume du kyste ou même dans certains cas de la faire disparaître.
Injections en vue d’obtenir un effet local dans les dermatoses
Dans la zone aseptisée, injecter 20 à 60 mg de suspension. Pour les cas où la zone des liaisons est plus importante, injecter plusieurs doses avec des quantités plus faibles (de l'ordre de 20 à 40) afin d'augmenter l'efficacité.
Pour un effet général
Lorsque l'on pratique une injection de DEPO-MEDROL par voie intramusculaire, la dose à injecter dépend de la gravité de l'infection et de son étendue. Si l'on veut un effet prolongé, la dose à injecter correspond à 7 fois la dose prise par la voie orale quotidienne. Il est impératif de prendre en compte l'âge et le poids corporel de l'individu.
Les injections intramusculaires de 80 à 120 mg peuvent avoir des effets allant de quelques jours à 2 semaines.
Chez les sujets souffrant de rhinite allergique (rhume des foins), une dose intramusculaire de 80 à120mg peut soulager les symptômes dans un délai de 6 heures, et ce soulagement peut durer de quelques jours à 3 semaines.
Surdosage
Le traitement du surdosage aigu consiste à prodiguer des soins de soutien et à soulager les symptômes. Pour éviter un surdosage chronique en présence d’une maladie grave nécessitant une corticothérapie continue, on peut réduire la posologie du corticostéroïde, pourvu que cet ajustement soit temporaire. En cas de surdosage, il faut savoir qu’il n’existe pas d’antidote spécifique de la méthylprednisolone.
La méthylprednisolone est dialysable.
Notes et références
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- « WHO Model List of Essential Medicines, 18th list », avril 2013.
Bibliographie et sources
- Medrol, methylprednisolone : dossier technique par Laboratoires Upjohn.
- Bertrand Wechsler, Corticoïdes et corticothérapie, Olivier Chosidow (Google Livre).
- http://webpeda.ac-montpellier.fr/wspc/ABCDORGA/Famille5/CORTICOIDES.htm.
- http://french.raw-steroid-powder.com/sale-3049002-methylprednisolone-99-pharmaceuticals-intermediate-steroid-hormone-cas-83-43-2.html.
- http://corine.bensimon.pagesperso-orange.fr/corticost%8eroides.html.
- http://udsmed.u-strasbg.fr/pharmaco/pdf/DCEM1_Pharmacologie_chapitre_23_Les_anti_inflammatoires_steroidiens.pdf.
- http://www.chu-rouen.fr/page/detail/fr/MSH_D_000893.
- http://www.chups.jussieu.fr/polys/pharmaco/poly/POLY.Chp.14.2.html.
- http://www.sciencedirect.com/science/article/pii/S0248866313000672.
- http://www.lecrat.org/article.php3?id_article=663.
- http://www.has-sante.fr/portail/upload/docs/application/pdf/2008-11/solumedrol_-_ct-5879.pdf.
- http://www.pfizer.ca/fr/our_products/products/monograph/179Fichier:Creative-Tail-test+tube.svg+Portail+de+la+chimie++Fichier:Star+of+life2.svg+Portail+de+la+médecine++Fichier:Coupe+d'Hygie.svg+Portail+de+la+pharmacie+++++++++++++++++++.
{kind=link}