Glycogénose type 3
La glycogénose type III est une maladie génétique du métabolisme des glucides de la famille des glycogénoses qui se manifeste par une carence en amylo-1,6-glucocidase, l'enzyme débranchante (en) du glycogène. Le glycogène est une molécule que le corps humain utilise pour stocker les glucides. La déficience en enzyme débranchante entraine une accumulation de glycogène anormal dans le foie, ainsi que parfois dans les muscles et plus rarement dans le cœur.
| Glycogénose type 3 Maladie de Cori Maladie de Forbes Dextrinose limite | ||
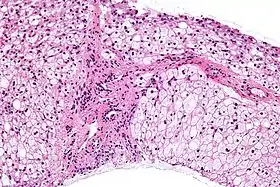 Micrographie d'une biopsie de foie présentant les caractéristiques d'une maladie de Cori. H&E stain (en). | ||
| Référence MIM | #232400 | |
|---|---|---|
| Transmission | Récessive | |
| Chromosome | 1q21.2 | |
| Gène | AGL | |
| Prévalence | 1 sur 100 000 naissances viables | |
| Liste des maladies génétiques à gène identifié | ||
Cette maladie est également connue sous les noms suivants :
- Maladie de Cori en hommage aux lauréats du prix Nobel 1947, Carl Cori et Gerty Cori
- Maladie de Forbes en hommage à l'américain Gilbert Burnett Forbes (1915-2003), qui a décrit les caractéristiques de cette maladie
- Dextrinose limite[1]
- Il est parfois possible de voir le nom de Maladie d'Illingworth-Cori-Forbes[2] en hommage aux précédents ainsi qu'à Barbara Illingworth, qui a travaillé avec Carl et Gerty Cori.
Prévalence
La glycogénose de type III est une maladie à transmission autosomique récessive. Bien qu'il n'existe pas de décompte précis, les études américaines et européennes montrent qu'elle apparait environ 1 fois toutes les 100 000 naissances viables. Cette prévalence s’élèverait à 1 pour 5 000 chez les Inuits d'Amérique du Nord et les juifs séfarades d'origine nord-africaine, voire à 1 pour 3 600 pour la population des îles Féroé[3].
Présentation
La glycogénose type III est divisée en au moins 4 sous-catégories :
- Le type IIIa (majoritaire) se manifeste par le déficit en enzyme débranchante dans les muscles et le foie[4]
- Les symptômes du type IIIb (15 % des cas) se restreignent au foie et n'ont pas d'impact sur le glycogène des muscles
- Le type IIIc (rare) se caractérise par la perte de l'activité débranchante dans les muscles mais pas dans le foie[5]
- Le type IIId (également rare) se caractérise par une mauvaise activité de glucosyltransferase (en) dans les muscles et le foie. Si l'enzyme déficiente est différente, les symptômes sont très proches de ceux de la type IIIa.
La maladie se manifeste le plus souvent pendant la petite enfance à travers les hypoglycémies et un retard de croissance. Un examen médical révèle souvent une hépatomégalie. Les symptômes musculaires (hypotonie et cardiomyopathie) apparaissent habituellement plus tard. Dans la majorité des cas, les symptômes hépatiques régressent à l'adolescence et seule une faible proportion des patients développe un cirrhose à l'âge adulte.
Traitement
À ce jour, il n'existe pas de remède à la maladie. Les traitements palliatifs peuvent être un régime apportant beaucoup de protéines pour faciliter la glycogénolyse et la néoglucogénèse, ainsi que des apports réguliers de glucides dont l’absorption est lente (maïs, blé, ...) pour éviter les hypoglycémies.
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Glycogen storage disease type III » (voir la liste des auteurs).
Liens externes
Références
- eMedicine The Continually Updated Clinical Reference
- Description de la maladie sur le site du téléthon
- eMedicine The Continually Updated Clinical Reference
- Lucchiari S, Fogh I, Prelle A, et al., « Clinical and genetic variability of glycogen storage disease type IIIa: seven novel AGL gene mutations in the Mediterranean area », Am. J. Med. Genet., vol. 109, no 3, , p. 183–90 (PMID 11977176, DOI 10.1002/ajmg.10347)
- Article paru sur nature.com intitulé "Glycogen Storage Disease Type III diagnosis and management guidelines"