Réaction de cyclisation de Nazarov
La réaction de cyclisation de Nazarov (souvent appelée simplement la cyclisation de Nazarov ) est une réaction chimique utilisée en chimie organique pour la synthèse de cyclopenténones . La réaction est généralement divisée en variantes classiques et modernes, en fonction des réactifs et des substrats utilisés. Elle a été découvert à l'origine par Ivan Nikolaevich Nazarov (1906-1957) en 1941 alors qu'il étudiait les réarrangements des cétones vinyliques allyliques[1] - [2].

Comme décrit à l'origine, la cyclisation de Nazarov implique l'activation d'une divinyl cétone en utilisant un promoteur tel qu'un acide de Lewis ou un acide protique en proportion stœchiométrique. L'étape clé du mécanisme de réaction implique une fermeture de cycle électrocyclique cationique 4π qui forme le produit sous la forme d'un dérivé de la cyclopenténone (voir mécanisme ci-dessous). Au fur et à mesure que la réaction s'est développée, des variantes impliquant des substrats autres que les divinyl cétones et des promoteurs autres que les acides de Lewis ont été classées sous le nom de cyclisation de Nazarov à condition qu'elles suivent une voie mécanique similaire.
Le succès de la cyclisation de Nazarov en tant qu'outil de synthèse organique découle de l'utilité et de la forte présence des cyclopenténones en tant que motifs dans les produits naturels (y compris la jasmone, les aflatoxines et une sous-classe de prostaglandines) et en tant qu'intermédiaires synthétiques utiles pour des synthèses totales. La réaction a été utilisée dans plusieurs synthèses totales et plusieurs articles ont été publiées[3] - [4] - [2] - [5] - [6] - [7].
Mécanisme
Le mécanisme de la réaction de cyclisation classique de Nazarov a d'abord été démontré expérimentalement par Shoppe comme une électrocyclisation intramoléculaire et est décrit ci-dessous. L'activation de la cétone par le catalyseur acide génère un cation pentadiényle, qui subit une électrocyclisation conrotatoire 4π thermiquement autorisée comme dicté par les règles de Woodward-Hoffman. Cela génère un cation oxyallyle qui subit une réaction d'élimination d'un β-hydrogène. Une tautomérisation ultérieure de l'énolate produit le dérivé de la cyclopenténone[8] - [9].
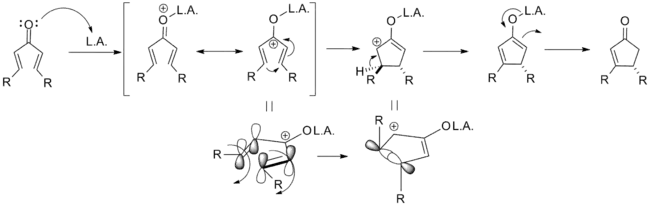
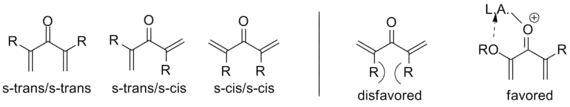
Comme indiqué ci-dessus, des variantes qui s'écartent de ce modèle sont connues. Ce qui désigne en particulier une cyclisation de Nazarov est la génération du cation pentadiényle suivie d'une fermeture électrocyclique du cycle en cation oxyallyle. Pour réaliser cette transformation, la molécule doit être dans la conformation s-trans/s-trans, plaçant les groupes vinyle dans une orientation appropriée. La propension du système à entrer dans cette conformation influence considérablement la vitesse de réaction et les substrats substitués en α ayant un nombre accru de conformère requis en raison de la souche allylique. La coordination d'un substituant α donneur d'électrons par le catalyseur peut également augmenter la vitesse de réaction en imposant cette conformation[3].
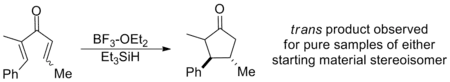
De même, la substitution en β dirigée vers l'intérieur restreint la conformation s-trans si sévèrement qu'il a été démontré que l' isomérisation EZ se produit avant la cyclisation sur une large gamme de substrats, produisant la trans cyclopenténone quelle que soit la configuration initiale. De cette façon, la cyclisation de Nazarov est un rare exemple de réaction péricyclique stéréosélective, alors que la plupart des électrocyclisations sont stéréospécifiques. L'exemple ci-dessous utilise de l'hydrure de triéthylsilyle pour piéger le cation oxyallyle afin qu'aucune élimination ne se produise[3]. (Voir cyclisations interrompues ci-dessous)
Le long de cette même réaction, les cétones allényl vinyliques du type étudié en détail par Marcus Tius de l'Université d'Hawaï montrent une accélération spectaculaire du taux d'élimination des β-hydrogènes, ce qui évite un grand nombre d'interaction stérique dans les conformères s-cis[6].

Cyclisations classiques de Nazarov
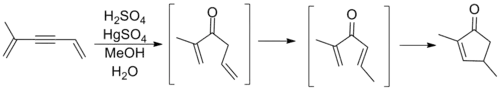
Bien que des cyclisations suivant le modèle général ci-dessus aient été observées avant l'implication de Nazarov, c'est son étude des réarrangements des cétones vinyl allyliques qui a marqué le premier examen majeur de ce processus. Nazarov a correctement estimé que l'oléfine allylique est isomérisée in situ pour former une divinyl cétone avant la fermeture du cycle du produit cyclopenténone. La réaction montrée ci-dessous implique une réaction d'oxymercuration d'alcyne pour générer la cétone requise[10].
La recherche impliquant la réaction a été relativement calme au cours des années suivantes, jusqu'au milieu des années 1980, lorsque plusieurs synthèses utilisant la cyclisation de Nazarov ont été publiées. Les étapes clés des synthèses du trichodiène et du nor-stéréépolide sont présentées ci-dessous, et l'on pense que ce dernier se déroule via une isomérisation inhabituelle d'alcyne-allène qui génère la divinyl cétone[11] - [12].

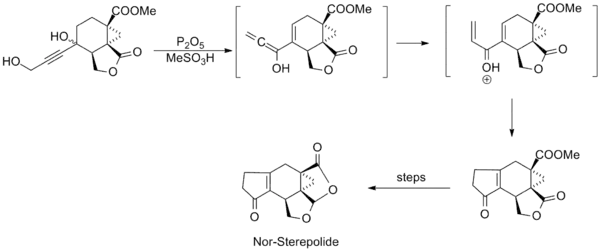
Lacunes
La version classique de la cyclisation de Nazarov souffre de plusieurs inconvénients que les variantes modernes tentent de contourner. Les deux premiers ne sont pas évidents d'après le mécanisme seul, mais indiquent les obstacles à la cyclisation; les trois derniers découlent de problèmes de sélectivité liés à l'élimination et à la protonation de l'intermédiaire[3].
- Des acides de Lewis ou protiques forts sont généralement nécessaires pour la réaction (par exemple, TiCl4, BF3, MeSO3H). Ces promoteurs ne sont pas compatibles avec les groupes fonctionnels sensibles, limitant la portée du substrat.
- Malgré la possibilité mécanistique de catalyse, plusieurs équivalents du promoteur sont souvent nécessaires pour effectuer la réaction. Cela limite l'économie atomique de la réaction.
- L'étape d'élimination n'est pas régiosélective ; si plusieurs β-hydrogènes sont disponibles pour l'élimination, divers produits sont souvent observés sous forme de mélanges. Ceci est hautement indésirable du point de vue de l'efficacité car une séparation difficile est généralement requise.
- L'élimination détruit un stéréocentre potentiel, diminuant l'utilité potentielle de la réaction.
- La protonation de l'énolate n'est parfois pas stéréosélective, ce qui signifie que les produits peuvent être formés sous forme de mélanges d'épimères .
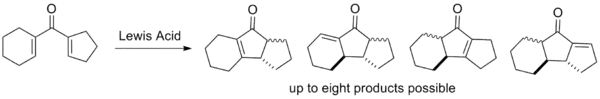
Variantes modernes
Les lacunes notées ci-dessus limitent l'utilité de la réaction de cyclisation de Nazarov sous sa forme canonique. Cependant, les modifications de la réaction axées sur la résolution de ses problèmes continuent d'être un domaine actif de la recherche universitaire. En particulier, la recherche s'est concentrée sur quelques domaines clés: rendre la réaction catalytique vis-à-vis du promoteur, effectuer la réaction avec des promoteurs plus doux pour améliorer la tolérance aux groupes fonctionnels, diriger la régiosélectivité de l'étape d'élimination et améliorer la stéréosélectivité globale. Celles-ci ont réussi à des degrés divers.
De plus, les modifications visaient à modifier la progression de la réaction, soit en générant le cation pentadiényle d'une manière peu orthodoxe, soit en faisant "intercepter" le cation oxyallyle de diverses manières. De plus, des variantes énantiosélectives de divers types ont été développées. Le volume considérable de littérature sur le sujet empêche un examen approfondi de ce domaine; des exemples clés sont donnés ci-dessous.
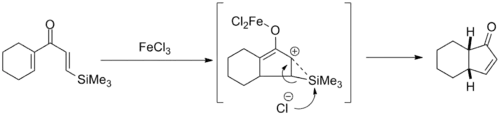
Cyclisation dirigée par silicium
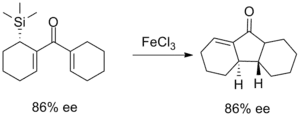
Les premiers efforts pour améliorer la sélectivité de la cyclisation de Nazarov ont profité de l'effet β-silicium pour diriger la régiosélectivité de l'étape d'élimination. Cette chimie a été largement développée par le professeur Scott Denmark de l'Université de l'Illinois à Urbana-Champaign au milieu des années 1980 et utilise des quantités stoechiométriques de trichlorure de fer pour favoriser la réaction. Avec les produits bicycliques, l'isomère cis a été sélectionné à des degrés divers[13].
La réaction de cyclisation de Nazarov dirigée par silicium a ensuite été utilisée dans la synthèse du produit naturel tel que le Silphinène, illustré ci-dessous. La cyclisation a lieu avant l'élimination de la fraction d'alcool benzylique, de sorte que la stéréochimie résultante du cycle nouvellement formé résulte de l'approche de l'alcène silylique en anti par rapport à l'éther[10].
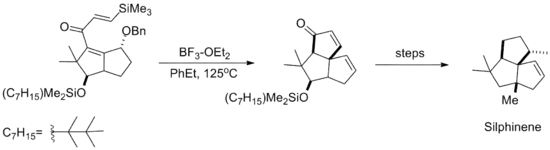
Polarisation
En se basant sur les effets des substituants compilés au cours de divers essais sur la réaction, le professeur Alison Frontier de l'Université de Rochester a développé un paradigme pour les cyclisations de Nazarov "polarisées" dans lesquelles des groupes électroattracteur et donneur d'électron sont utilisés pour améliorer la sélectivité globale de la réaction. La création d'un vinyle nucléophile et d'un vinyle électrophile efficaces dans le substrat permet une activation catalytique avec du triflate de cuivre et une élimination régiosélective. De plus, le groupe attracteur d'électrons augmente l'acidité du α-proton, permettant la formation sélective du trans-α-épimère par équilibrage[14].
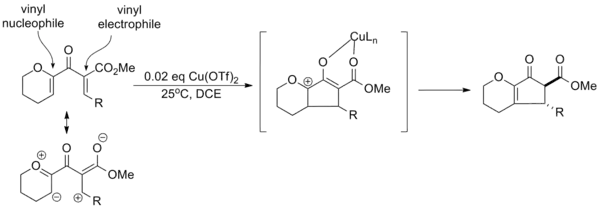
Il est souvent possible de réaliser l'activation catalytique en utilisant un groupe donneur ou électro-attracteur seul, bien que l'efficacité de la réaction (rendement, temps de réaction, etc.) soit généralement plus faible.
Génération alternative de cations
Par extension, tout cation pentadiényle quelle que soit son origine est susceptible de subir une cyclisation de Nazarov. Il y a eu un grand nombre d'exemples publiés où le cation requis est obtenu par une variété de réarrangements[3]. Un tel exemple implique l'ouverture du cycle cationique catalysée par l'argent du dichloro-cyclopropanes allyliques. Le sel d'argent facilite la perte de chlorure par précipitation de chlorure d'argent insoluble[15].
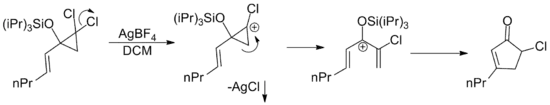
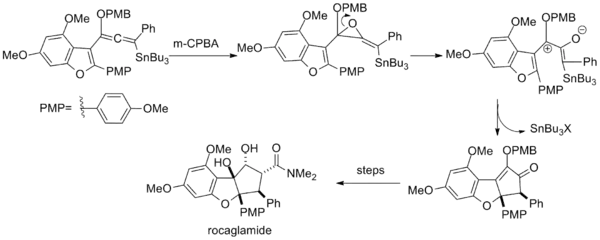
Dans la synthèse totale du rocaglamide, l'époxydation d'un vinylalcoxyallényl stannane génère également un cation pentadiényle via l'ouverture du cycle de l'époxyde résultant[16].
Cyclisations interrompues
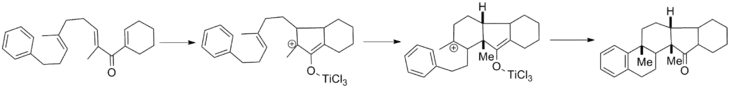
Une fois la cyclisation terminée, un cation oxyallyle se forme. Comme discuté en détail ci-dessus, le cours typique de cet intermédiaire est l'élimination suivie d'une tautomérisation d'énolate. Cependant, ces deux étapes peuvent être interrompues par divers nucléophiles et électrophiles, respectivement. Le piégeage des cations oxyallyliques a été largement développé par Fredrick G. West de l'Université de l'Alberta et son article couvre le domaine[17]. Le cation oxyallyle peut être piégé avec des hétéroatomes et des carbones nucléophiles et peut également subir des cycloadditions cationiques avec divers partenaires attachés. Ci-dessous, une réaction en cascade dans laquelle le piégeage de cations successifs génère un noyau pentacyclique en une seule étape avec une diastéréosélectivité complète[18].
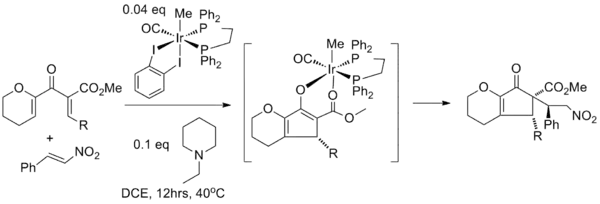
Le piégeage d'énolate avec divers électrophiles est nettement moins courant. Dans une étude, la cyclisation de Nazarov est associée à une réaction de Michael utilisant un catalyseur à l'iridium pour initier l'addition conjugué nucléophile de l'énolate au β-nitrostyrène. Dans cette réaction en tandem, le catalyseur à l'iridium est requis pour les deux réaction : il agit comme l'acide de Lewis dans la cyclisation de Nazarov et à l'étape suivante, le groupe nitro du nitrostyrène se coordonne d'abord à l'iridium dans un échange de ligands avec l'atome d'oxygène de l'ester carbonylique avant que l'addition de Michael a lieu à la face opposée du groupe R[19].
Variantes énantiosélectives
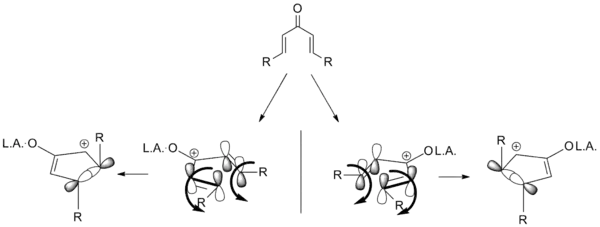
Le développement d'une cyclisation énantiosélective de Nazarov est un ajout souhaitable au répertoire des réactions de cyclisation de Nazarov. À cette fin, plusieurs variantes ont été développées en utilisant des auxiliaires chiraux et des catalyseurs chiraux. Des cyclisations diastéréosélectives sont également connues, dans lesquelles des stéréocentres existants dirigent la cyclisation. Presque toutes les tentatives sont basées sur l'idée de torquosélectivité ; sélectionner une direction pour que les groupes de vinyle "tournent" à leur tour et définissent la stéréochimie comme indiqué ci-dessous[3].
Les cyclisations de Nazarov dirigées par silicium peuvent ainsi présenter une diastéréosélectivité induite. Dans l'exemple ci-dessous, le groupe silyle agit pour diriger la cyclisation en empêchant l'alcène distant de tourner "vers" via une interaction stérique défavorable. De cette façon, le silicium agit comme un auxiliaire sans trace. (Le matériel de départ n'est pas énantiopur mais la rétention de l'excès énantiomérique suggère que l'auxiliaire dirige la cyclisation[3].)
Les substrats allényles de Tius peuvent présenter un transfert de chiralité axiale à tétraédrique si des allènes énantiopurs sont utilisés. L'exemple ci-dessous génère un diosphenpol chiral avec un rendement de 64% et un excès énantiomérique de 95%[3].
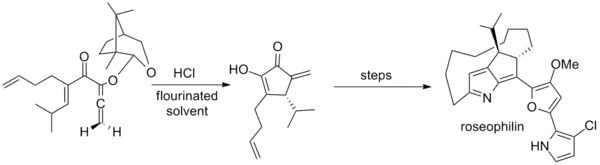
Tius a en outre développé un auxiliaire à base de camphre pour les allènes achiraux qui a été utilisé dans la première synthèse asymétrique de la roseophiline. L'étape clé utilise un mélange inhabituel d'hexafluoro-2-propanol et de trifluoroéthanol comme solvant[3] - [20].
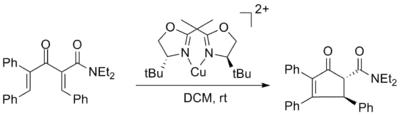
Varinder Aggarwal a signalé la première cyclisation chirale asymétrique favorisée par un acide chiral de Lewis et utilisé des complexes de ligands de cuivre (II) bisoxazoline avec jusqu'à 98% ee. L'excès énantiomérique n'a pas été affecté par l'utilisation de 50% en mole du complexe de cuivre mais le rendement a été considérablement diminué[3].
Réactions associées
Les extensions de la cyclisation de Nazarov sont généralement également regroupées sous le même nom. Par exemple, une cétone insaturée α-β, γ-δ peut subir une cyclisation conrotatoire cationique similaire qui est généralement appelée réaction de cyclisation iso-Nazarov[21]. D' autres extensions ont été nommées avec des noms similaires, y compris la cyclisation homo-Nazarov et la cyclisation vinylogues de Nazarov[22] - [23].
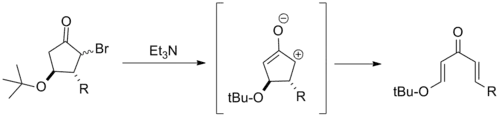
Réaction de rétro-Nazarov
Parce qu'ils surstabilisent le cation pentadiényle, les substituants donneurs d'électrons β entravent souvent gravement la cyclisation de Nazarov. À partir de cela, plusieurs ouvertures de cycles électrocycliques de β-alcoxy cyclopentanes ont été signalées. Celles-ci sont généralement appelées réactions de rétro-cyclisation de Nazarov[3].
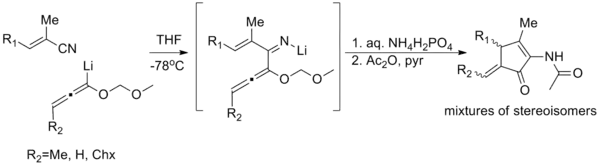
Réaction Imino-Nazarov
Les analogues azotés de la réaction de cyclisation de Nazarov (appelés réactions de cyclisation imino-Nazarov) ont peu d'exemples; il existe un exemple de cyclisation généralisée d'imino-Nazarov rapporté (illustré ci-dessous)[24], et plusieurs réactions d'iso-imino-Nazarov dans la littérature[25] - [26]. Même ceux-ci ont tendance à souffrir d'une mauvaise stéréosélectivité, de faibles rendements ou d'une portée limitée. La difficulté provient de la sur-stabilisation relative du cation pentadiényle par don d'électrons, empêchant la cyclisation[27].
Voir également
- Pauson–Khand reaction
- Electrocyclization
- Cyclopentenone
- Merrilactone A
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Nazarov cyclization reaction » (voir la liste des auteurs).
- (en) « The Nazarov Cyclization », Synform, (lire en ligne).
- Karl L. Habermas, Scott E. Denmark et Todd K. Jones, « The Nazarov Cyclization », dans Organic Reactions, John Wiley & Sons, Inc., (ISBN 0-471-26418-0, lire en ligne), p. 1–158
- (en) Alison J. Frontier et Christina Collison, « The Nazarov cyclization in organic synthesis. Recent advances », Tetrahedron, vol. 61, no 32, , p. 7577–7606 (DOI 10.1016/j.tet.2005.05.019, lire en ligne, consulté le )
- (en) Christiane Santelli-Rouvier et Maurice Santelli, « The Nazarov Cyclisation », Synthesis, vol. 1983, no 06, , p. 429–442 (ISSN 0039-7881 et 1437-210X, DOI 10.1055/s-1983-30367, lire en ligne, consulté le )
- Denmark, S.E. (1991), Paquette, L.A. (ed.), "The Nazarov and Related Cationic Cyclizations", Comprehensive Organic Synthesis, Oxford: Pergamon Press, 5: 751–784, doi:10.1016/b978-0-08-052349-1.00138-4, (ISBN 9780080523491)
- (en) Marcus A. Tius, « Some New Nazarov Chemistry », European Journal of Organic Chemistry, vol. 2005, no 11, , p. 2193–2206 (ISSN 1434-193X et 1099-0690, DOI 10.1002/ejoc.200500005, lire en ligne, consulté le )
- (en) Hélène Pellissier, « Recent developments in the Nazarov process », Tetrahedron, vol. 61, no 27, , p. 6479–6517 (DOI 10.1016/j.tet.2005.04.014, lire en ligne, consulté le )
- (en) C. W. Shoppee et Ruth E. Lack, « Intramolecular electrocyclic reactions. Part I. Structure of ‘bromohydroxyphorone’: 3-bromo-5-hydroxy-4,4,5,5-tetramethylcyclopent-2-enone », J. Chem. Soc. C, vol. 0, no 10, , p. 1346–1349 (ISSN 0022-4952, DOI 10.1039/J39690001346, lire en ligne, consulté le )
- (en) C. W. Shoppes et B. J. A. Cooke, « Intramolecular electrocyclic reactions. Part II. Reactions of 1,5-di-phenylpenta-1,4-dien-3-one », Journal of the Chemical Society, Perkin Transactions 1, , p. 2271 (ISSN 0300-922X et 1364-5463, DOI 10.1039/p19720002271, lire en ligne, consulté le )
- Kürti, L. et Czakó, B., Strategic Applications of Named Reactions in Organic Synthesis, Burlington, MA, Elsevier Academic Press, , 304–305 p. (ISBN 9780124297852, lire en ligne)
- (en) Kenn E. Harding et Katherine S. Clement, « A highly stereoselective, convergent synthesis of (.+-.)-trichodiene », The Journal of Organic Chemistry, vol. 49, no 20, , p. 3870–3871 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo00194a054, lire en ligne, consulté le )
- (en) Yoshitsugu Arai, Kunio Takeda, Katsutada Masuda et Toru Koizumi, « SYNTHESIS OF (±)-NOR-STEREPOLIDE », Chemistry Letters, vol. 14, no 10, , p. 1531–1534 (ISSN 0366-7022 et 1348-0715, DOI 10.1246/cl.1985.1531, lire en ligne, consulté le )
- (en) S. E. Denmark et T. K. Jones, « Silicon-directed Nazarov cyclization », Journal of the American Chemical Society, vol. 104, no 9, , p. 2642–2645 (ISSN 0002-7863, DOI 10.1021/ja00373a055, lire en ligne, consulté le )
- (en) Wei He, Xiufeng Sun et Alison J. Frontier, « Polarizing the Nazarov Cyclization: Efficient Catalysis under Mild Conditions », Journal of the American Chemical Society, vol. 125, no 47, , p. 14278–14279 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja037910b, lire en ligne, consulté le )
- (en) Tina N. Grant et F. G. West, « A New Approach to the Nazarov Reaction via Sequential Electrocyclic Ring Opening and Ring Closure », Journal of the American Chemical Society, vol. 128, no 29, , p. 9348–9349 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja063421a, lire en ligne, consulté le )
- Malona, J.A.; Cariou, K.; Frontier, A.J. (2009), "Nazarov Cyclization Initiated by Peracid Oxidation: The Total Synthesis of (±)-Rocaglamide", J. Am. Chem. Soc., 131 (22): 7560–7561, doi:10.1021/ja9029736, PMC 2732401,
- (en) Tina N. Grant, Curtis J. Rieder et Frederick G. West, « Interrupting the Nazarov reaction: domino and cascade processes utilizing cyclopentenyl cations », Chemical Communications, no 38, , p. 5676 (ISSN 1359-7345 et 1364-548X, DOI 10.1039/b908515g, lire en ligne, consulté le )
- (en) John A. Bender, Atta M. Arif et F. G. West, « Nazarov-Initiated Diastereoselective Cascade Polycyclization of Aryltrienones 1 », Journal of the American Chemical Society, vol. 121, no 32, , p. 7443–7444 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja991215f, lire en ligne, consulté le )
- (en) Mesfin Janka, Wei He, Inga E. Haedicke et Frank R. Fronczek, « Tandem Nazarov Cyclization−Michael Addition Sequence Catalyzed by an Ir(III) Complex », Journal of the American Chemical Society, vol. 128, no 16, , p. 5312–5313 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja058772o, lire en ligne, consulté le )
- (en) Paul E. Harrington et Marcus A. Tius, « A Formal Total Synthesis of Roseophilin: Cyclopentannelation Approach to the Macrocyclic Core », Organic Letters, vol. 1, no 4, , p. 649–652 (ISSN 1523-7060 et 1523-7052, DOI 10.1021/ol990124k, lire en ligne, consulté le )
- (en) Michael E. Jung et Dongwon Yoo, « Unprecedented Rearrangement of a 4-Alkoxy-5-bromoalk-2-en-1-ol to a Cyclopentenone via an Iso-Nazarov Cyclization Process », The Journal of Organic Chemistry, vol. 72, no 22, , p. 8565–8568 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo071104w, lire en ligne, consulté le )
- (en) Filippo De Simone, Julien Andrès, Riccardo Torosantucci et Jérôme Waser, « Catalytic Formal Homo-Nazarov Cyclization », Organic Letters, vol. 11, no 4, , p. 1023–1026 (ISSN 1523-7060 et 1523-7052, DOI 10.1021/ol802970g, lire en ligne, consulté le )
- (en) Curtis J. Rieder, Karl J. Winberg et F. G. West, « Cyclization of Cross-Conjugated Trienes: The Vinylogous Nazarov Reaction », Journal of the American Chemical Society, vol. 131, no 22, , p. 7504–7505 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja9023226, lire en ligne, consulté le )
- (en) Marcus A Tius, Chester C Chu et Raquel Nieves-Colberg, « An imino Nazarov cyclization », Tetrahedron Letters, vol. 42, no 13, , p. 2419–2422 (DOI 10.1016/S0040-4039(01)00201-5, lire en ligne, consulté le )
- Se-Ho Kim et Jin Kun Cha, « Synthetic Studies Toward caphalotaxine: Functionalization of Tertiary N-Acylhemiaminals by Nazarov Cyclization », Synthesis, vol. 2000, no 14, , p. 2113–2116 (DOI 10.1055/s-2000-8711, lire en ligne, consulté le )
- (en) Paolo Larini, Antonio Guarna et Ernesto G. Occhiato, « The Lewis Acid-Catalyzed Nazarov Reaction of 2-( N -Methoxycarbonylamino)-1,4-pentadien-3-ones », Organic Letters, vol. 8, no 4, , p. 781–784 (ISSN 1523-7060 et 1523-7052, DOI 10.1021/ol053071h, lire en ligne, consulté le )
- (en) Douglas A. Smith et Charles W. Ulmer, « Effects of Substituents in the 3-Position on the [2 + 2] Pentadienyl Cation Electrocyclization 1 », The Journal of Organic Chemistry, vol. 62, no 15, , p. 5110–5115 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo9703313, lire en ligne, consulté le )