Halogénation électrophile aromatique
Une halogénation électrophile aromatique est une réaction organique de type substitution électrophile aromatique qui permet d'ajouter des substituants de type halogène sur un composé aromatique :
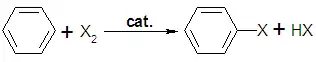
Quelques composés aromatiques, tels que le phénol, peuvent réagir par cette réaction sans catalyseur, mais la plupart des dérivés benzéniques avec des substituants moins réactifs nécessitent une catalyse par un acide de Lewis. Les catalyseurs typiques de cette réaction sont AlCl3, FeCl3, FeBr3 et ZnCl2. La réaction se produit en formant un complexe fortement électrophile qui va attaquer le cycle aromatique.
Mécanisme réactionnel
Le mécanisme réactionnel de la bromation ou de la chloration du benzène est le même. Le bromure ferrique et le chlorure ferrique étant inactivés en présence d'eau (la simple humidité de l'air suffit), il est nécessaire de faire la réaction dans un montage hermétiquement clos avec une verrerie passée au préalable à l'étuve. En général, on produit le catalyseur in situ, en ajoutant la limaille de fer au brome ou au chlore. Le mécanisme de la réaction est le suivant :
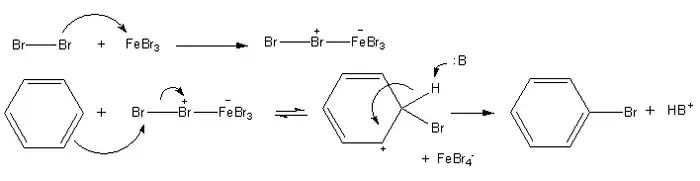
Dans un premier temps, le dibrome réagit avec le bromure ferrique pour former un zwitterion où l'un des atomes de brome devient chargé positivement et électrophile. L'un des doublets libre du benzène vient alors attaquer l'atome de brome terminal, formant un cation arénium. Finalement, un ion bromure peut agir comme une base et vient arracher l'hydrogène supplémentaire, permettant de former le bromobenzène.
Le mécanisme pour l'iodation est légèrement différent : le diiode (I2) est traité par un oxydant, tel que l'acide nitrique, pour obtenir un iode électrophile (2 I+).
L'halogénation des composés aromatiques diffère de celle des alcènes, ces derniers n'ayant pas besoin de catalyse par un acide de Lewis. La formation d'un ion arénium résulte d'une perte temporaire d'aromaticité, ce qui provoque une hausse de l'énergie d'activation comparé aux alcènes.
Utilisation
Si le cycle contient un substituant activateur fort, tel que -OH, -OR ou une amine, un catalyseur n'est pas nécessaire. On peut citer par exemple la bromation du p-crésol[1] :
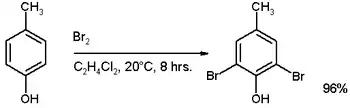
Cependant, si on utilise un catalyseur avec un excès de brome, on forme un dérivé tribromé.
L'halogénation des phénols est plus rapide dans les solvants polaires, de par la dissociation du phénol formant un ion phénoxyde, plus susceptible de subir une attaque électrophile puisque plus riches en électrons.
La chloration du toluène par le dichlore sans catalyseur requiert un solvant polaire tel que l'acide acétique. La sélectivité ortho/para est faible[2] :
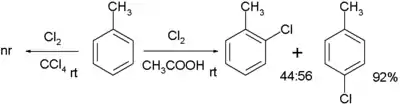
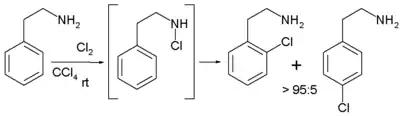
Au contraire, aucune réaction ne se produit si le solvant est remplacé par le tétrachlorométhane. Par contre, si le réactif est une 2-phényléthylamine, il est possible d'utiliser un solvant relativement apolaire, avec régiosélectivité exclusive ortho, du fait de la formation d'un intermédiaire chloramine, ce qui rend l'étape suivante de la réaction intramoléculaire.
L'érythrosine, un colorant alimentaire, peut être synthétisé par iodation d'un autre colorant, la fluorescéine :
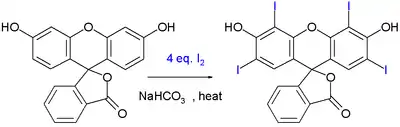
Cette réaction est catalysée par le bicarbonate de sodium[3].
Notes et références
- (en) A. Sankaranarayanan et S. B. Chandalia, « Process Development of the Synthesis of 3,4,5-Trimethoxytoluene », Org. Process Res. Dev., vol. 10, no 3, , p. 487–492 (ISSN 1083-6160, DOI 10.1021/op0502248).
- (en) J. L. O'Connell, J. S. Simpson et al., « Aromatic chlorination of ω-phenylalkylamines and ω-phenylalkylamides in carbon tetrachloride and α,α,α-trifluorotoluene », Org. Biomol. Chem., vol. 4, no 14, , p. 2716-2723 (ISSN 1477-0520, PMID 16826296, DOI 10.1039/b605010g).
- (en) J. V. McCullagh et K. A. Daggett, « Synthesis of Triarylmethane and Xanthene Dyes Using Electrophilic Aromatic Substitution Reactions », J. Chem. Educ., vol. 84, no 11, , p. 1799 (ISSN 0021-9584, DOI 10.1021/ed084p1799).
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Electrophilic halogenation » (voir la liste des auteurs).
Articles connexes
- Substitution électrophile aromatique
- Fluoration électrophile
- Halogénation