Cladistique
La cladistique[1] - [2] (ou systématique phylogénétique[3] - [4] - [5]) est la théorie des clades et des cladogrammes[6] (du grec ancien κλάδος / kládos, « branche »), et de la reconstruction des relations de parenté entre les êtres vivants[7]. Un clade (groupe monophylétique) est un groupe dont tous les membres sont plus apparentés entre eux qu'avec n'importe quel autre groupe, et un cladogramme (arbre phylogénétique)[8] est une hiérarchie de clades. Le cladogramme spécifie des relations de degrés de parenté entre les taxons qu'il classifie[9]. Dans le cadre de la théorie de l'évolution, les relations de degré de parenté entre taxons sont expliquées par l'existence d'ancêtres communs (i.e. le fait que deux taxons soient plus proches parents entre eux que d'un troisième implique que les deux premiers descendent d'un ancêtre commun exclusif). La théorie cladistique a été initialement présentée dans les années 1950 par l'entomologiste allemand Willi Hennig (1913-1976).
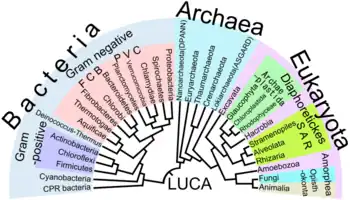
L'analyse cladistique[10] fournit, au sein de la théorie cladistique, la méthode permettant la reconstruction des cladogrammes sur la base d'états de caractères dérivés partagés, également appelés synapomorphies. En cela, elle permet la reconstruction phylogénétique dans le cadre de la théorie cladistique. L'analyse cladistique utilise la synapomorphie, acquise par un ancêtre et hérité par tous ses descendants, afin de proposer des hypothèses de clades[11] (Caractère phénotypique). Les caractères peuvent en cladistique inclure des connaissances morphologiques, génétiques, biochimiques ou encore comportementales. La cladistique est encore aujourd'hui utilisée en phylogénétique[12] où les analyses sont réalisées à l'aide de programmes informatiques[13]. Elle est également utilisée en taxonomie pour définir des taxons reflétant l'évolution des espèces[14] - [15] - [16].
Clades et cladogrammes
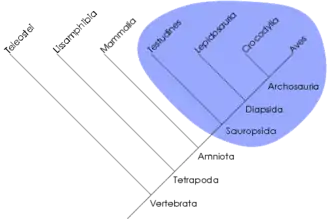
Un clade est un groupe d'organismes monophylétique, c'est-à-dire dont tous les membres, si différents soient-ils devenus, descendent d'un même groupe-ancêtre commun. Ce concept ne s'oppose pas à celui de grade évolutif, rapprochant des organismes sur d'autres critères (par ex. ressemblance générale, somme de modifications adaptatives semblables alors que ces organismes ont des ancêtres différents : c'est le cas des « rapaces », nom vernaculaire sans valeur phylétique, désignant des oiseaux prédateurs, d'origines diverses, mais ayant tous développé, indépendamment les uns des autres, des becs crochus et des serres). La cladistique qualifie les grades de « groupes paraphylétiques » ou « polyphylétiques » selon que le rapprochement est effectué sur la base de plésiomorphies ou d'homoplasies. Certains grades peuvent toutefois être monophylétiques. Par exemple, les algues forment un grade polyphylétique, les reptiles forment un grade paraphylétique et les mammifères forment un grade monophylétique.
On parle aussi parfois de groupe monophylétique pour désigner un clade, Hennig ayant voulu redéfinir ce terme pour en exclure la paraphylie, ce qui a jeté une certaine confusion terminologique[17].
Un clade peut aussi être défini comme un ensemble d'organismes phylogénétiquement plus proches les uns des autres qu'ils ne le sont d'aucun autre organisme. Par exemple, les reptiles ne forment pas un clade car certains (les crocodiliens) sont plus apparentés aux oiseaux (formant ainsi le clade archosauriens) qu'aux autres reptiles. Ainsi, un clade correspond à une unité évolutive.
Par définition le taxon est l'unité des classifications scientifiques du vivant. Dans le cadre du cladisme, tous les taxons sont des clades et tous les clades sont des taxons, contrairement à la taxonomie numérique ou à la systématique évolutionniste où un grade paraphylétique peut être un taxon valide.
Il est parfois fait mention d'« espèce ancestrale » pour désigner une espèce représentante d'un clade. Cependant, une telle espèce, si elle existait, serait nécessairement définie sur la base d'états de caractères plésiomorphes par rapport à ses descendants. Elle correspondrait alors à la définition d'un groupe paraphylétique et ne serait pas reconnue comme unité évolutive par la théorie cladistique. Dans ce cadre théorique, l'ancêtre est, premièrement, hypothétique et, deuxièmement, n'est pas caractérisable en tant que taxon. « Non caractérisable » ne signifie pas « non identifiable » : il est donc possible de dresser un morphotype ancestral.
Paraphylie
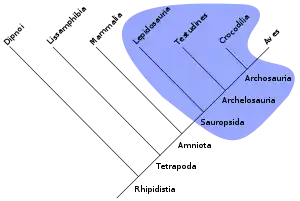
La notion de paraphylie s'applique à un groupe défini sur le partage d'un ou de plusieurs états de caractères plésiomorphes. Les taxons de ce genre de groupes ne constituent pas une totalité de descendance puisque les organismes portant les états apomorphes en sont exclus. La théorie cladistique ne reconnaît pas la pertinence de groupes tels que les reptiles, les poissons, les invertébrés, les procaryotes, etc. Au sein d'un groupe paraphylétique, les taxons ne sont pas forcément plus apparentés entre eux qu'à d'autres taxons. Par exemple, les tétrapodes sont un clade au sein d'un groupe plus général comprenant aussi les poissons. Certains poissons comme le cœlacanthe sont plus proches des tétrapodes qu'ils ne le sont d'autres poissons tels le requin. Ainsi, le terme poisson désigne un groupe paraphylétique.
Le cladogramme étant le résultat d'un test des états de caractères, c'est lui qui indique si un groupe est monophylétique ou paraphylétique. Il est donc important de justifier le choix des états de caractères considérés, en amont de l'analyse, apomorphes ou plésiomorphes (le test des états de caractères ne concernant que les états apomorphes). Pour cela, il existe plusieurs critères dont celui du groupe externe (le plus utilisé), le critère ontogénétique et à moindre mesure les critères paléontologique et chorologique. Soient deux états a et b d'un même caractère dont la relation peut être schématisée par a↔b en l'absence de justification sur l’apomorphie ou la plésiomorphie de l'un ou de l'autre. Le schéma a→b indiquerait a comme étant plésiomorphe et b apomorphe. Le schéma b→a indiquerait le contraire.
Homologie versus homoplasie

On distingue deux types de ressemblances, l'homologie et l'homoplasie. Selon la définition la plus courante, des états de caractères dits homologues sont hérités d'un ancêtre commun. Pour quelques auteurs, la relation entre états est appelée homologie. Deux états homologues sont non seulement ressemblants, mais surtout phylogénétiquement « identiques ». À l'inverse, l'homoplasie, terme introduit par Lankester en 1870[18], désigne des états de caractères non hérités d'un ancêtre commun. Des états dits homoplastiques sont donc ressemblants mais ne sont pas phylogénétiquement « identiques ». Savoir distinguer la source de ces ressemblances est une tâche compliquée. Plusieurs critères permettent de proposer le statut « homologues » pour ces ressemblances, le plus utilisé étant le « principe des connexions » d'Étienne Geoffroy Saint-Hilaire[19] (ensuite repris par Richard Owen[20] - [21]) appelé aussi « identité des connexions » : deux organes sont homologues si, quelles que soient leurs formes et/ou fonctions, ils ont les mêmes connexions avec d'autres organes. Ce principe peut aisément s'étendre aux séquences moléculaires et sous-tend la pratique d'alignement des séquences.
Ce principe ne permet que de formuler des hypothèses dites d'homologie primaire. En effet rien ne nous assure que les états de caractère supposés homologues le sont effectivement. La cladistique offre le cadre théorique pour tester de telles hypothèses. Il s'agit d'un test de congruence selon Hennig ou de parcimonie. Le principe de congruence (ou de parcimonie) cherche à maximiser le nombre d'hypothèses d'homologie compatibles entre elles afin de minimiser le nombre d'hypothèses surnuméraires (ou ad hoc), une hypothèse ad hoc étant une hypothèse d'homoplasie (pouvant recevoir quantité d'explications la rendant non testable). Le cladogramme reconstruit est le résultat de la maximisation de la congruence parmi les hypothèses d'homologie. Une hypothèse non rejetée par le test est appelée « homologie secondaire ».
À l'inverse, une hypothèse d'homologie rejetée par le test permet d'attribuer le statut d'homoplasie aux états en question. N'importe quelle explication permettrait de comprendre l'origine des homoplasies. Deux explications principales sont souvent discutées : la convergence évolutive et la réversion. La convergence indique qu'une ressemblance est apparue plusieurs fois indépendamment. La réversion est interprétée comme étant la perte secondaire d'un état de caractère, c'est-à-dire le retour à un état ressemblant à l'état plésiomorphe. Par exemple tous les Vertébrés ne possèdent pas de membres pairs. Or l'ensemble des Vertébrés concernés par cette absence ne forme pas un groupe monophylétique (ni monophylétique ni paraphylétique). On dit par exemple des Gymnophiones ou des Serpents qu'ils ont « perdu » ces membres pairs. L'homoplasie n'étant pas un caractère hérité par un ancêtre commun, elle ne nous renseigne pas sur les relations de parenté. Un groupe identifié sur la base d'une homoplasie est appelé groupe polyphylétique.
Analyse cladistique
Différentes méthodes de reconstruction phylogénétique existent en cladistique. Ces méthodes se basent sur deux principes méthodologiques définis par Hennig : la règle de regroupement et le principe auxiliaire ("grouping rule" et "auxiliary principle")[7] - [22]. La règle de regroupement énonce que l'existence d'un clade ne peut être justifié que par l'existence d'une synapomorphie. Le principe auxiliaire, lui, énonce qu'une relation phylogénétique doit toujours être considérée comme vraie, à moins d'être contredite par d'autres relations.
Plusieurs méthodes se réclament de l'analyse cladistique :
- l'argumentation hennigienne[7]
- la parcimonie (dite standard)[23] - [24]
- l'optimisation directe[25]
- l'analyse à trois éléments (3ia)[26] - [27]
- la compatibilité[28]
Si les méthodes de phylogénie statistique (voir phylogénétique moléculaire) telles que le maximum de vraisemblance ou l'inférence bayésienne utilisent le cadre théorique cladistique (reconstruction de l'évolution par la recherche des groupes monophylétiques uniquement), le fait qu'elles soient des méthodes cladistiques ou non est sujet à controverse[29] - [30].
Argumentation hennigienne
Hennig, le fondateur de la théorie cladistique, n'a jamais utilisé ces méthodes aujourd'hui assistées par ordinateur. Il parlait de méthode d'argumentation. Aujourd'hui une phylogénie n'est que très rarement acceptée sans analyse algorithmique pour l'appuyer.
Cette procédure non automatisée consiste à proposer une phylogénie (ou schéma d'argumentation) sur la base d'arguments pour chaque clade, les arguments étant les états de caractères explicitement donnés par l'auteur.
Parcimonie
Selon la méthode de parcimonie standard, l'économie d'hypothèse touche au nombre de pas évolutifs. L'arbre le plus court (c'est-à-dire l'arbre avec le moins de pas évolutifs) est l'arbre représentant l'hypothèse phylogénétique la plus acceptable. Le comptage des pas peut être influencé par la considération que l'on a des convergences et/ou réversions, d'où l'existence de plusieurs écoles méthodologiques :
- La parcimonie de Wagner (convergences et réversions sont acceptées).
- La parcimonie de Camin-Sokal (les convergences sont admises mais pas les réversions).
- La parcimonie de Dollo (les réversions sont admises mais pas les convergences).
Interprétation du cladogramme en parcimonie
L'attribution des états de caractères aux taxons est représentée, le plus généralement, dans un tableau appelé matrice taxons-caractères. Voici la matrice hypothétique dans laquelle, pour chaque caractère "x", l'état plésiomorphe est noté x et l'état apomorphe x'.
| caractère | taxon A | taxon B | taxon C | taxon D | taxon E | taxon F |
|---|---|---|---|---|---|---|
| Caractère "a" | a | a | a' | a' | a' | a' |
| Caractère "b" | b | b | b' | b | b | b |
| Caractère "c" | c' | c | c | c | c' | c' |
| Caractère "d" | d | d' | d' | d' | d | d' |
| Caractère "e" | e | e | e | e | e | e |
Soient les taxons A, B, C, D, E et F. On considère l'arbre suivant : (A(B((C, D)(E, F)))). Ici R={A+B+C+D+E+F} : c'est la racine ou le nœud qui contient tout. H= {B+C+D+E+F} G= {C+D+E+F} = {I, J} avec I= {C+D} et J= {E+F}. Les nœuds internes H, G, I et J sont des taxons au même titre qu'A, B, C, D, E et F.
L'histoire parcimonieuse des états de caractères est représentée par des barres bleues où x→x' indique le passage de l'état x à x' et x'→x le passage de l'état x' à l'état x. Le passage d'un état à un autre est appelé transformation ou pas évolutif.
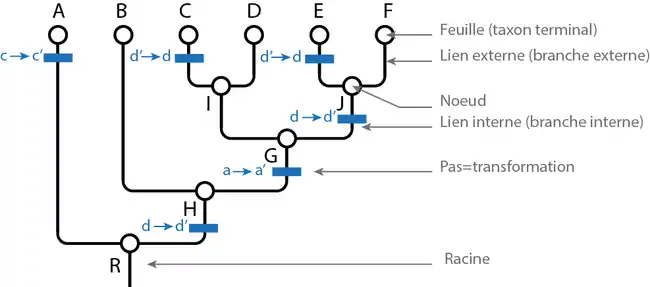
Ici, l'état a' est commun aux taxons C, D, E, et F; c'est donc une synapomorphie de G.
L'état b' n'apparaît que sur le seul taxon terminal C. C'est donc une autapomorphie de C. Cet état ne renseigne pas sur les relations de parenté de C avec les autres taxons.
L'état e est commun à tous les taxons, c'est donc une symplésiomorphie.
L'état c' apparaît deux fois dans l'arbre. Le passage de c à c' coûte donc deux pas évolutifs. L'état c' est une homoplasie (il n'est donc pas interprété comme hérité d'un ancêtre commun).
Le passage de l'état d à d' est suivi d'un autre passage inverse de d' à d. Cette deuxième transformation est généralement interprétée comme une réversion.
Le coût total des transformations est de 6 pas évolutifs.
On dit de deux taxons qu'ils sont groupes-frères quand ils sont plus proches entre eux que de n'importe quel troisième taxon. Ici par exemple, C et D sont groupes-frères, ainsi que B et G ou encore J et I.
Un cladogramme est un graphe particulier, appelé hiérarchie en mathématiques. Une hiérarchie est équivalente à un emboîtement de classes. Chaque classe correspond à un nœud du graphe. Au sens phylogénétique, un nœud ou une classe est un taxon. Ici les taxons C et D sont inclus dans une classe I. Tous les taxons (terminaux ou inclusifs) sont inclus dans R : la définition de la racine est d'être la classe incluant toutes les autres classes.
Un graphe hiérarchique peut être représenté par un diagramme de Venn pour faire apparaître l'emboîtement des classes. L'image ci-dessous représente le diagramme de Venn correspondant au cladogramme utilisé dans l'exemple précédent.

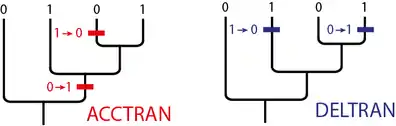
Dans certains cas il n'y a aucun élément pour savoir si l'homoplasie est une réversion ou une convergence, on parle alors d'interprétation ambiguë[31]. Le choix réversion ou convergence est arbitraire[32]. L'hypothèse ACCTRAN (Accelerated transformation) favorise les réversions. L'hypothèse DELTRAN (Delayed transformation) favorise les convergences. L'image ci-dessous l'illustre pour deux arbres ayant la même répartition des caractères.
À titre d'exemple, l'absence de fenêtre antéorbitaire chez les crocodiles actuels est considéré comme une réversion (elle était présente chez leurs ancêtres putatifs) ; les topologies similaires du bassin des oiseaux et des ornitischiens, ou encore les formes hydrodynamiques des delphinidés et de la plupart des lamniformes sont considérées comme des convergences.
Enracinement et groupe externe
Le principe dit du « groupe externe » ou « extra-groupe » est généralement utilisé pour enraciner un arbre phylogénétique, c'est-à-dire désigner le nœud correspondant à la classe racine (contenant toutes les autres classes)[33]. On estime que tout état de caractère observé en dehors du groupe interne est plésiomorphe pour le groupe interne. Au contraire, tout état propre à des taxons du groupe d'étude est considéré apomorphe.
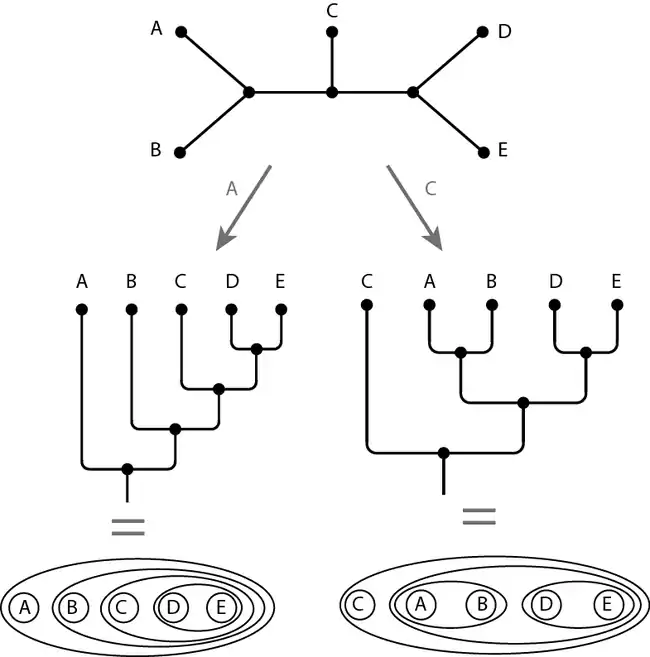
Une des méthodes mettant en œuvre ce principe introduit un taxon du groupe externe dans l'analyse : par exemple un téléostéen pour une étude phylogénétique des tétrapodes ; un céphalopode ou un bivalve pour une étude phylogénétique des gastéropodes. Il faut veiller à ce que les caractères des groupes externe et interne soient comparables, ce qui incite à choisir un groupe externe relativement proche du groupe d'étude. Dans l'image ci-dessous, l'arbre de départ n'est pas enraciné alors que les deux arbres ci-dessous (sous forme de graphe ou de diagramme de Venn) sont enracinés, c'est-à-dire orientés grâce aux groupes externes A ou C.
Les relations de parenté et le sens des transformations de caractères ne peuvent être inférées qu'à partir d'un arbre enraciné (autrement dit : du cladogramme).
On remarque que le choix du groupe externe (ici A ou C) modifie la topologie de l'arbre.
Il arrive que la méthode décrite ci-dessus outrepasse le principe énoncé. En effet, le taxon du groupe externe introduit dans l'analyse ne présente pas nécessairement la totalité des caractères sous l'état plésiomorphe.
D'autres méthodes d'enracinement proposent d'appliquer ce principe pour chaque caractère, en comparant le groupe d'étude à plusieurs taxons du groupe externe. Ces taxons ne sont alors pas introduits dans l'analyse :
- soit un taxon hypothétique est reconstruit ;
- soit les caractères sont polarisés en amont de l'analyse (dans le cas d'une représentation par matrice) ;
- soit les caractères sont hiérarchisés en amont de l'analyse (dans le cas d'une représentation par hiérarchie).
Optimisation directe
Selon la méthode d'optimisation directe, l'économie d'hypothèses se situe en amont de l'analyse, dans le codage même des caractères. Techniquement, proposer les caractères conduisant à l'arbre parcimonieux s'effectue en même temps que la recherche de cet arbre (c'est-à-dire lors du comptage des pas). Dans un cadre d'analyse moléculaire, cette méthode ne nécessite pas d'alignement des séquences préalablement à l'analyse. Les nucléotides de séquences alignées sont équivalents au codage d'états de caractères identiques. Ici, l'alignement (ou le codage) est donc effectué au fur et à mesure de l'analyse de manière à optimiser le nombre de pas. L'arbre obtenu par optimisation directe est théoriquement plus court qu'un arbre obtenu par une autre méthode d'alignement[34].
Compatibilité
Selon la méthode de compatibilité, l'économie d'hypothèses concerne le nombre de caractères permettant d'éviter les homoplasies. On dit de tels caractères qu'ils sont mutuellement compatibles. L'ensemble des caractères mutuellement compatibles formera une clique. L'arbre retenu sera construit à partir de la clique la plus importante. Il sera donc dépourvu d'homoplasie.
Analyse à trois éléments
Selon la méthode d'analyse à trois éléments ou 3ia (Three item analysis, parfois aussi appelée TTS pour Three-Taxon Statements), l'économie d'hypothèses concerne la congruence des relations : minimiser l'incongruence, ou maximiser la congruence.
L'analyse à trois éléments décompose les caractères en hypothèses relationnelles à trois éléments spécifiant que deux taxons sont plus proches entre eux que d'un troisième. Ces hypothèses minimales sont appelées 3is pour three item statements ou « assertions à trois éléments », d'où le nom de la méthode. Cela implique d'avoir accès aux hypothèses relationnelles (ou de parenté) sur les caractères : les caractères sont donc logiquement représentés sous la forme d'arbres racinés, ou autrement dit, de graphes hiérarchiques. Un caractère consiste à attribuer les états d'une structure morpho-anatomique aux taxons de l'étude après avoir indiqué la relation (hiérarchique) entre ces états.
L'arbre phylogénétique reconstruit l'est à partir de l'ensemble le plus grand de 3is compatibles entre eux.
La 3ia a d'abord été utilisée pour la biogéographie historique et a ensuite été utilisée pour l'étude des taxons dans le cadre théorique de la cladistique. Cette méthode n'utilise pas, pour l'analyse, de taxon du groupe externe. Le systématicien applique en amont de l'analyse le principe extra-groupe, entre autres, pour désigner les états informatifs des caractères, c'est-à-dire les états permettant de regrouper des taxons.
Très peu d'études ont comparé la performance de l'analyse à trois éléments à celle de la parcimonie. La plus récente[35] a trouvé que la 3ia donnait une excellente puissance et un taux d'erreurs (clades artéfactuels) intermédiaires entre celui de la parcimonie avec états ordonnés (donnant le moins d'erreurs) et celui de la parcimonie non-ordonnée (donnant le plus d'erreurs).
Critiques
Dans les organismes à reproduction sexuelle, le tri de lignées incomplet peut avoir pour résultat des arbres phylogénétiques contradictoires les uns avec les autres, en fonction des gènes qui sont étudiés[36].
Notes et références
- « Encyclopédie Larousse en ligne - cladistique », sur larousse.fr (consulté le ).
- Ernst Mayr et Willi Hennig (trad. Martin S. Fischer et Pascal Tassy), Analyse cladistique : le débat Mayr-Hennig de 1974, Paris, Éditions matériologiques, (lire en ligne)
- Encyclopædia Universalis, « CLADISTIQUE ou SYSTÉMATIQUE PHYLOGÉNÉTIQUE », sur Encyclopædia Universalis (consulté le )
- (en) John L. Gittleman, « Phylogeny | biology », Encyclopædia Britannica, (lire en ligne, consulté le )
- (en) Richard M. Kliman, Encyclopedia of evolutionary biology, , 2132 p. (ISBN 978-0-12-800049-6, 012800049X et 9780128098127, OCLC 926743072, lire en ligne), p. 344
- (en) Ian J. Kitching, Peter L. Forey, David Williams et Christopher Humphries, Cladistics : The Theory and Practice of Parsimony Analysis, Oxford University Press, , 228 p. (ISBN 978-0-19-850138-1, lire en ligne)
- (en) Willi Hennig (trad. D. Dwight Davis et Rainer Zangerl), Phylogenetic Systematics, Urbana, Chicago, London, University of Illinois Press, , 263 p. (lire en ligne)
- « Phylogénie (Pascal Tassy) – e-systematica », sur e-systematica.org (consulté le )
- Pierre Darlu et Pascal Tassy, La reconstruction phylogénétique (lire en ligne)
- « Analyse cladistique (Pascal Tassy) – e-systematica », sur e-systematica.org (consulté le )
- (en) Chi-Chun Ho, Susanna K. P. Lau et Patrick C. Y. Woo, « Romance of the three domains : how cladistics transformed the classification of cellular organisms », Protein & Cell, vol. 4, no 9, , p. 664–676 (ISSN 1674-800X et 1674-8018, PMID 23873078, PMCID PMC4875529, DOI 10.1007/s13238-013-3050-9, lire en ligne, consulté le )
- (en) « The Future of Phylogenetic Systematics », Cambridge Core, (DOI 10.1017/cbo9781316338797, lire en ligne, consulté le )
- (en) Swofford, « PAUP*: phylogenetic analysis using parsimony, version 4.0b10 », http://paup.phylosolutions.com/, (lire en ligne, consulté le )
- Angiosperm Phylogeny Group
- (en) Kevin de Queiroz, « The PhyloCode and the Distinction between Taxonomy and Nomenclature », Systematic Biology, vol. 55, no 1, , p. 160–162 (ISSN 1076-836X et 1063-5157, DOI 10.1080/10635150500431221, lire en ligne, consulté le )
- (en) P. Bouchet, Yu I. Kantor, A. Sysoev et N. Puillandre, « A new operational classification of the Conoidea (Gastropoda) », Journal of Molluscan Studies, vol. 77, no 3, , p. 273–308 (ISSN 0260-1230, DOI 10.1093/mollus/eyr017, lire en ligne, consulté le )
- Dubois, A. (1986). À propos de l’emploi controversé du terme “monophylétique”: nouvelles propositions. Bulletin mensuel de la Société linnéenne de Lyon, 55, 248-254.
- (en) E. Ray Lankester, "On the use of the term Homology in modern Zoology and the distinction between Homogenetic and Homoplastic agreements", Annals and Magazine of Natural History, Vol.6, No.31, 1870, p.34-43. DOI 10.1080/00222937008696201
- Étienne Geoffroy Saint-Hilaire, Philosophie anatomique : Des organes respiratoires sous le rapport de la détermination et de l'identité de leurs pièces osseuses, Méquignon-Marvis, Paris, 1818. lire en ligne sur Gallica
- (en) R. Owen, Lectures on Comparative Anatomy and Physiology of the Invertebrate Animals, Longman, Brown, Green and Longmans, London, 1843.
- (en) R. Owen, On the Archetype and Homologies of the Vertebrate Limb, John Van Voorst, London, 1848.
- E. O. Wiley, The Compleat cladist : a primer of phylogenetic procedures, Lawrence, Kan. : Museum of Natural History, University of Kansas, (lire en ligne)
- (en) « Kluge and Farris, 1969, Syst. Zool., 18 | Amphibian Species of the World », sur research.amnh.org (consulté le )
- James S. Farris, « Methods for Computing Wagner Trees », Systematic Zoology, vol. 19, no 1, , p. 83–92 (DOI 10.2307/2412028, lire en ligne, consulté le )
- (en) « Exploring the Behavior of POY, a Program for Direct Optimization of Molecular Data », Cladistics, vol. 17, no 1, , S60–S70 (ISSN 0748-3007, DOI 10.1006/clad.2000.0159, lire en ligne, consulté le )
- R. Zaragüeta-Bagils & E. Bourdon, Three-item analysis: Hierarchical representation and treatment of missing and inapplicable data, Comptes Rendus Palevol, Volume 6, Issues 6-7, November 2007, Pages 527-534
- (en) Gareth Nelson et Norman I. Platnick, « THREE-TAXON STATEMENTS: A MORE PRECISE USE OF PARSIMONY? », Cladistics, vol. 7, no 4, , p. 351–366 (ISSN 0748-3007 et 1096-0031, DOI 10.1111/j.1096-0031.1991.tb00044.x, lire en ligne, consulté le )
- (en) « A mathematical foundation for the analysis of cladistic character compatibility », Mathematical Biosciences, vol. 29, nos 1-2, , p. 181–187 (ISSN 0025-5564, DOI 10.1016/0025-5564(76)90035-3, lire en ligne, consulté le )
- (en) James S. Farris, « Popper: not Bayes or Rieppel », Cladistics, vol. 29, no 3, , p. 230–232 (ISSN 0748-3007, DOI 10.1111/cla.12010, lire en ligne, consulté le )
- (en) W. C. Wheeler et K. M. Pickett, « Topology-Bayes versus Clade-Bayes in Phylogenetic Analysis », Molecular Biology and Evolution, vol. 25, no 2, , p. 447–453 (ISSN 0737-4038 et 1537-1719, DOI 10.1093/molbev/msm274, lire en ligne, consulté le )
- M.G.G. de Pinna, Concepts and tests of homology in the cladistic paradigm, Cladistics, 1991.En ligne (en)
- I. Agnarson & J. A. Miller Is ACCTRAN better than DELTRAN?, Cladistics, 2008 En ligne (en)
- V. Barriel & P. Tassy Rooting with Multiple Outgroup: Consensus Versus Parsimony, Cladistics, 1998
- (en) Filip Högnabba, Botanical Museum, Finnish Museum of Natural History, University of Helsinki, « Direct optimization » [PDF] (consulté le ).
- Grand A, Corvez A, Duque Velez LM, Laurin M. (2013). Phylogenetic inference using discrete characters: performance of ordered and unordered parsimony and of three-item statements. Biological Journal of the Linnean Society, 110 (4): 914–930.
- (en) Jeffrey Rogers et Richard A. Gibbs, « Comparative primate genomics: emerging patterns of genome content and dynamics », Nature Reviews Genetics, vol. 15, no 5, , p. 347–359 (PMID 24709753, PMCID 4113315, DOI 10.1038/nrg3707)
Voir aussi
Bibliographie
- P. Tassy, La renaissance de la systématique, 2001 En ligne (fr)
- G. Lecointre & H. Le Guyader, Classification phylogénétique du vivant, 1re édition, Belin, 2001 ; 3e édition, Belin, 2006.
- P. Tassy, L'arbre à remonter le temps, Christian Bourgois, Paris, 1991.
- C. Patterson, Morphological characters and homology, in K.A. Joysey et A.E. Friday (éd.), Problems in Phylogenetic Reconstruction, Londres, Academic Press, 1982.
- K. de Queiroz et J.A. Gauthier, « Phylogenetic taxonomy », Annual Review of Ecology and Systematics no 23, 1992, p. 449–480.
- D.L. Swofford, G.J. Olsen, P.J. Waddell et D.M. Hillis, Phylogenetic inference, in D.M. Hillis, C. Moritz et B.K. Mable (éd.), Molecular Systematics, Sunderland (Massachusetts), Sinauer Associates, 1996.
- E.O. Wiley, Phylogenetics : The Theory and Practice of Phylogenetic Systematics.-, New York, Wiley Interscience, 1981.