Chimie organique physique
La chimie organique physique, discipline baptisée par Louis Plack Hammett en 1940, est une branche de la chimie organique qui se concentre sur la relation entre structure chimique et réactivité en appliquant les méthodes de la chimie physique à l'étude des molécules organiques.
La chimie organique physique étudie notamment les vitesses des réactions chimiques organiques, la stabilité chimique relative, les intermédiaires réactionnels, les états de transition, et les effets non covalents de la solvatation et des interactions moléculaires sur la réactivité chimique. De telles études fournissent des cadres théoriques et pratiques pour comprendre comment des changements de structure en solution impactent le mécanisme et la vitesse d'une réaction organique donnée. Les spécialistes de chimie organique physique utilisent des approches théoriques et expérimentales pour comprendre ces problèmes fondamentaux de la chimie organique : thermodynamique classique et statistique, mécanique quantique, chimie numérique, spectroscopie (RMN), spectrométrie (spectrométrie de masse) et cristallographie.
Portée
Le champ d'application de la chimie organique physique est vaste : électrochimie, photochimie, polymérisation et chimie supramoléculaire, chimie bioorganique, enzymologie, biochimie, mais également dans l'industrie avec la chimie des procédés, le génie chimique, la science des matériaux et les nanotechnologies, et l'industrie pharmaceutique.
Structure chimique et thermodynamique
Thermochimie
Les chimistes organiciens emploient les outils de la thermodynamique pour étudier la stabilité de la liaison chimique et l'énergie des systèmes chimiques. Cela inclut notamment des expériences pour mesurer ou déterminer l'enthalpie (ΔH), l'entropie (ΔS), et l'enthalpie libre (ΔG) d'une réaction. Les chimistes peuvent également utiliser des analyses mathématiques variées comme le graphe de Van't Hoff pour calculer ces valeurs.
Des constantes empiriques comme l'énergie de dissociation, l'enthalpie standard de formation (ΔHf°) et de combustion (ΔHc°), sont utilisées pour prédire la stabilité des molécules et la variation d'enthalpie (ΔH) au cours d'une réaction. Pour des molécules complexes, la valeur de l'enthalpie de formation peut être inconnue mais peut être estimée en décomposant la molécule concernée en fragments moléculaires d'enthalpies de formation connues. Cette méthode est souvent rapportée à la théorie d'incrément de groupe de Benson (Benson group increment theory BGIT), en l'honneur du chimiste Sidney Benson qui passa sa carrière à développer ce concept[1] - [2].
La thermochimie des intermédiaires réactionnels - carbocations, carbanions et radicaux - est également un sujet d'intérêt pour les spécialistes de chimie organique physique. Les données d'incrément de groupe sont disponibles pour les systèmes radicaux. La stabilité des carbocations et des carbanions peut être estimée en utilisant respectivement les affinités ioniques hybrides et les valeurs de pKa.
Analyse conformationnelle
Une des méthodes principales pour estimer la stabilité chimique et énergétique est l'analyse conformationnelle. Les spécialistes de chimie organique physique utilisent l'analyse conformationnelle pour évaluer les différents types de contraintes présentes dans une molécule et prédire les produits de réaction[3].
En plus de prédire la stabilité moléculaire, l'analyse conformationnelle prédit également les produits de réaction. Un exemple fréquemment cité de l'utilisation de l'analyse conformationnelle est la réaction d'élimination bimoléculaire (E2). Lors de cette réaction, le nucléophile attaque le site qui est en position antipériplanaire par rapport au groupe partant. Une analyse orbitalaire de ce phénomène suggère que cette conformation fournit le meilleur recouvrement entre les électrons de l'orbitale liante σ de la liaison R-H qui est en train d'être attaquée par le nucléophile, et l'orbitale antiliante σ* de la liaison R-X qui est en train de se rompre[4]. En exploitant cet effet, l'analyse conformationnelle peut être utilisée afin de créer des molécules possédant une meilleure réactivité.
Les processus physiques qui donnent lieu à des barrières de rotation de liaison sont complexes, et ces barrières ont été largement étudiées par des méthodes expérimentales et théoriques[5] - [6] - [7]. Des articles scientifique ont traité le sujet de la prédominance des contributions stériques, électrostatiques, et d'hyperconjugaison aux barrières rotationnelles dans l'éthane, le butane, et d'autres molécules substituées[8].
Interactions non covalentes
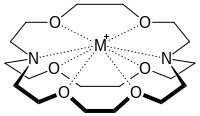
Les chimistes étudient les interactions non covalentes intramoléculaires et intermoléculaires dans les molécules afin d'évaluer leur réactivité. Ces interactions incluent notamment la liaison hydrogène, des interactions électrostatiques entre molécules chargées, des interactions dipôle-dipôle, et les liaisons halogènes. En outre, l'hydrophobie due à l'association de composés organiques dans l'eau est une interaction électrostatique non covalente d'intérêt pour les chimistes. L'origine physique précise de l'hydrophobie est liée à de nombreuses interactions complexes. L'étude des interactions non covalentes permet également de comprendre les liaisons et la coopérativité des édifices supramoléculaires et des composés macrocycliques tels que les éthers couronne ou les cryptands.
Acido-basicité
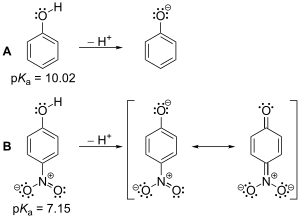
L'acido-basicité relève de la chimie organique physique. Les chimistes organiciens s'intéressent surtout aux acides/bases de Brønsted qui échangent un proton, et aux acides/bases de Lewis qui échangent un électron. Les chimistes utilisent une série de facteurs, provenant de la chimie physique - électronégativité/induction, force de liaison, mésomérie, hybridation, aromaticité et solvatation - pour prédire les acidités et basicités relatives.
La théorie HSAB est utilisée pour prédire les interactions moléculaires et le sens de réaction. En général, les interactions entre molécules de même type sont favorables. Cela signifie que les acides durs réagiront de préférence avec les bases dures, et les acides mous avec les bases molles. Le concept d'acides et de bases durs est souvent exploité dans la synthèse de complexes de coordination.
Cinétique
Les spécialistes de chimie organique physique utilisent les bases mathématiques de la cinétique chimique pour étudier les vitesses de réaction et leur mécanisme. Contrairement à la thermodynamique qui traite de la stabilité relative des produits et des réactifs (ΔG°) et leur concentration à l'équilibre, la cinétique traite, elle, de l'énergie d'activation (ΔG‡)- qui est la différence d'enthalpie libre entre la structure du réactif et celle de l'état de transition - d'une réaction, ce qui permet au chimiste d'étudier le procédé d'équilibre. Certains principes venant des mathématiques comme le postulat de Hammond, le principe de Curtin-Hammett et la théorie de la réversibilité microscopique sont souvent appliqués à la chimie organique. Les chimistes jouent sur le conflit entre contrôle thermodynamique et contrôle cinétique pour avoir un produit de réaction plutôt qu'un autre.
Lois de vitesse
La cinétique chimique est utilisée pour déterminer la loi de vitesse d'une réaction. La loi de vitesse fournit une relation quantitative entre l'avancement d'une réaction chimique et les concentrations ou pressions des espèces chimiques en jeu. Les lois de vitesse sont déterminées par des mesures expérimentales et ne peuvent en général pas être déduites de l'équation de réaction. La vitesse de réaction déterminée expérimentalement correspond à la stœchiométrie de la structure de l'état de transition relativement à la structure de l'état fondamental. La détermination de la loi de vitesse a été historiquement accomplie en suivant la concentration d'un réactif pendant la réaction par analyse gravimétrique, mais est aujourd'hui presque exclusivement faite par des techniques spectroscopiques rapides et sans ambiguïtés. Dans la plupart des cas, la détermination de la loi de vitesse est simplifiée en ajoutant un large excès de tous les réactifs sauf un.
Catalyse
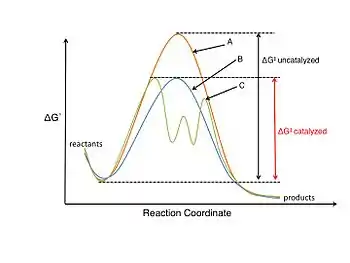
L'étude de la catalyse et des réactions catalytiques est un domaine très important de la chimie organique physique. Un catalyseur participe à la réaction chimique mais n'est pas consommé au cours de celle-ci. Un catalyseur abaisse l'énergie d'activation (ΔG‡), ce qui permet d'augmenter la vitesse de la réaction soit en stabilisant l'état de transition soit en déstabilisant un intermédiaire réactionnel clé.
Notes et références
- (en) N. Cohen et Benson, S. W., « Estimation of heats of formation of organic compounds by additivity methods », Chemical Reviews, vol. 93, no 7, , p. 2419–2438 (DOI 10.1021/cr00023a005)
- (en) Sidney W. Benson, Cruickshank, F. R., Golden, D. M., Haugen, Gilbert R., O'Neal, H. E., Rodgers, A. S., Shaw, Robert et Walsh, R., « Additivity rules for the estimation of thermochemical properties », Chemical Reviews, vol. 69, no 3, , p. 279–324 (DOI 10.1021/cr60259a002)
- (en) Francis A. Carey, Organic Chemistry, Boston, MA, USA, McGraw-Hill, , 7th éd. (ISBN 978-0-07-304787-4)
- (en) Neil S. Isaacs, Physical Organic Chemistry, Harlow, ESS, ENG, 2nd, , 877 p. (ISBN 0-582-21863-2)
- Yirong Mo et Gao, Jiali, « Theoretical Analysis of the Rotational Barrier of Ethane », Accounts of Chemical Research, vol. 40, no 2, , p. 113–119 (DOI 10.1021/ar068073w)
- Shubin Liu, « Origin and Nature of Bond Rotation Barriers: A Unified View », The Journal of Physical Chemistry A, vol. 117, no 5, , p. 962–965 (DOI 10.1021/jp312521z)
- Shubin Liu et Govind, Niranjan, « Toward Understanding the Nature of Internal Rotation Barriers with a New Energy Partition Scheme: Ethane and-Butane », The Journal of Physical Chemistry A, vol. 112, no 29, , p. 6690–6699 (DOI 10.1021/jp800376a)
- Takuhei Yamamoto, Chen, Pi-Yu, Lin, Guangxin, Błoch-Mechkour, Anna, Chen, Pi-Yu, Chen, Pi-Yu, Chen, Pi-Yu, Chen, Pi-Yu, Chen, Pi-Yu et Chen, Pi-Yu, « Synthesis and rotation barriers in 2, 6-Di-( -anisyl) anisole », Journal of Physical Organic Chemistry, vol. 25, no 10, , p. 878–882 (DOI 10.1002/poc.2939)