Angiœdème bradykinique
L’angiœdème bradykinique est une maladie génétique rare, qui se caractérise par des épisodes récurrents de gonflements (œdèmes) situés au niveau du visage, des muqueuses et des organes internes, parfois susceptibles de mettre en jeu le pronostic vital.
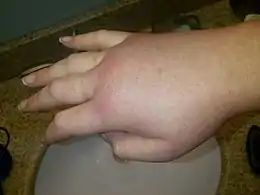
| Spécialité | Hématologie |
|---|
| CISP-2 | B99 |
|---|---|
| CIM-10 | D84.1 (ILDS D84.110) |
| CIM-9 | 277.6 |
| OMIM | 106100 |
| DiseasesDB | 1821 |
| MedlinePlus | 001456 |
| eMedicine | 1048994 |
| MeSH | D054179 |
| Patient UK | Hereditary-angio-oedema |
![]() Mise en garde médicale
Mise en garde médicale
Étymologie
- œdème : gonflement,
- angio- : vaisseau (sanguin)
Nomenclature
Selon Orphanet, l’angiœdème bradykinique est également dénommé angio-œdème héréditaire (AOH), angiœdème à kinines, angiœdème non histaminique ou œdème angioneurotique (OAN)[1] - [2].
Le terme « œdème angioneurotique héréditaire » est désuet, car on supposait qu’il était purement lié au stress suscité par un traumatisme émotionnel et / ou des névroses. Cette image imprécise de l’angiœdème bradykinique a conduit de nombreux malades à être qualifiés à tort d’instables mentaux ; on refusait souvent de traiter leurs symptômes qui étaient atrocement douloureux, notamment en cas de gonflement à l’intérieur de l’appareil gastro-intestinal, de la vessie et des organes reproducteurs. La médecine moderne a depuis lors discrédité cette théorie, retirant ainsi le terme « neurotique » de son nom[3].
Types
Il existe trois types d'angiœdème bradykinique[4], correspondant à des mutations différentes sur le gène de protéine :
- Type 1 – taux réduits de C1INH (85 %[5]) ;
- Type 2 – taux normaux de C1INH mais fonction réduite (15 %) ;
- Type 3 – anomalie non détectable de C1INH, survient dans un mode dominant lié à X, affectant en conséquence essentiellement les femmes ; il peut être exacerbé par la grossesse et l'utilisation de contraceptifs oraux (initialement décrit par Bork et al. en 2000, fréquence exacte incertaine).
Épidémiologie
La prévalence de l'angiœdème bradykinique est estimée entre un cas sur 10 000 et un cas sur 50 000 environ[6]. On estime à 50 000 personnes le nombre de patients atteints d'angiœdème bradykinique en Europe. Pour la France, l'estimation se situe autour de 1 000 malades. La plupart du temps, les premiers symptômes apparaissent vers l'âge de 10 ou 20 ans. L’angiœdème bradykinique concerne les femmes et les hommes à la même fréquence.
La fréquence est cependant probablement sous-estimée[7] - [8].
Causes
Il existe quelques formes acquises d'angiœdème bradykinique, d'origine allergique[9] - [10] et éventuellement médicamenteuse (allergie à l'hydroxychloroquine par exemple)[11]
Sinon la maladie est héréditaire à transmission autosomique dominante mais ne se déclare pas forcément ; un défaut génétique induit alors un déficit en inhibiteur de la C1 estérase (C1-INH), dont le gène est SERPING1[12]. Soit l’organisme produit une quantité trop faible de cette protéine (angiœdème de type 1), soit cette protéine n’est pas fonctionnelle (angiœdème de type 2)[13]. Les angiœdème bradykinique de type 1 sont cinq à six fois plus fréquents que les type 2[14]. Il a été décrit plus de 200 mutations causales de la maladie[15] - [16]. La transmission est de type autosomal dominant : chaque enfant d' un patient atteint aura un risque sur deux d'être lui aussi atteint, le taux de mutation de novo est de 1/4 : tous les patients n'ont pas un de leurs parents atteint[17].
Le C1-INH joue un rôle régulateur dans deux systèmes de réaction vitaux dans l’organisme : le système de contact de la coagulation sanguine et le système du complément du système immunitaire. Dans le cas de l’angiœdème bradykinique, un déficit en C1-INH dans les deux systèmes peut contribuer à la formation d’œdème ; le peptide bradykinine semble toutefois jouer le rôle central dans le système de contact. La libération de bradykinine intervient à la fin d’une cascade de réactions, qui est déclenchée par l’organisme en réponse à une lésion. Le peptide fait en sorte qu’une quantité accrue de liquide passe hors des vaisseaux dans les tissus, ce qui entraîne la formation d’œdèmes. Dans le même temps, il dilate les vaisseaux et déclenche les contractions des muscles lisses, qui provoquent les crampes et les douleurs. Normalement, la libération de bradykinine dépend du taux de kallikréine qui est limitée par l’action de C1-INH[18] ; en cas de déficit en C1-INH, une quantité de bradykinine sensiblement plus élevée que nécessaire est libérée.
Le déficit en C1-INH joue également en diminuant la concentration en C2 et C4, facteurs du système du complément. La baisse de ces facteurs n'a pas un rôle très clair dans la maladie mais constitue un marqueur diagnostic.
Les inhibiteurs de l'enzyme de conversion utilisés pour abaisser la pression artérielle peuvent également déclencher de sévères crises d’œdèmes[19], en raison de leur effet sur le métabolisme de la bradykinine[20]. Les crises peuvent également être favorisées par la prise d'une contraception orale ou d'un traitement substitutif de la ménopause[21] ainsi que par la présence d'Helicobacter pylori dans l'estomac[22]. Dans de nombreux cas, le déficit en C1-INH est dû à des causes inconnues (angiœdème idiopathique).
Les maladies auto-immunes peuvent également provoquer un déficit en C1-INH. Dans ces cas là, l’affection est appelée angiœdème bradykinique acquis.
Tableau clinique
Les premières crises débutent durant l'enfance, entre 8 et 12 ans[14]. La fréquence, de celles-ci est variable, en moyenne de 8 à 9 crises par an[23]. Elles sont rares avant 1 an et tendent à s'exacerber à l’adolescence[5].
Il n’existe souvent aucune cause directe identifiable, bien que des traumatismes légers et autres stimuli (stress par exemple) puissent être à l’origine des crises[24], ce qui fait qu'un traitement prophylactique est poposée avant la réalisation d'un acte médical agressif. Généralement, aucune démangeaison ni urticaire ne sont associées aux crises, étant donné qu’il ne s’agit pas d’une réaction allergique. Un œdème évolue progressivement sur 12 à 36 heures, puis régresse spontanément en 2 à 5 jours. Ces crises sont souvent précédées de prodromes (signes avant-coureurs) à type de fatigue, rashs cutanés ou douleurs musculaires[25].
Les œdèmes cutanés provoquent souvent des rougeurs et des douleurs, mais pas de démangeaisons. Ils affectent le plus souvent la peau ou les muqueuses du visage, les extrémités ou les parties génitales et peuvent défigurer.
Les gonflements au niveau des voies aériennes supérieures (larynx, nez, langue) sont particulièrement dangereux, car ils peuvent provoquer une suffocation s’ils ne sont pas traités à temps, le délai entre le début des symptômes et la suffocation pouvant être très court (quelques dizaines de minutes)[26]. L'angiœdème du pharynx et du larynx se manifeste comme un œdème de Quincke.
Les formes digestives sont souvent confondus avec ceux d’une colique abdominale ou d’une appendicite. Ils se caractérisent par des douleurs abdominales pouvant être accompagnées de nausées ou d'une diarrhée. Il peut exister une ascite[27]. Les crises gastriques peuvent durer de 1 à 5 jours en moyenne, et peuvent nécessiter une hospitalisation pour une prise en charge efficace de la douleur et hydratation. Les crises abdominales sont connues pour provoquer une augmentation significative de la numération leucocytaire des patients, habituellement de l’ordre de 13 à 30 000. À mesure que les symptômes commencent à régresser, la numération commence elle aussi à diminuer lentement, se normalisant dès lors que la crise cède.
L’angiœdème bradykinique provoque aussi un gonflement à divers autres endroits, la plupart du temps, au niveau des membres, des parties génitales, du cou, de la gorge et du visage. La douleur associée à ces gonflements varie de désagréable à atroce, en fonction de la localisation et de la sévérité.
Il est impossible de prévoir à quel endroit et quand se produira le prochain œdème. En moyenne, la plupart des personnes atteintes d’angiœdème bradykinique ont une crise par mois, Cependant, chez certains patients, l’angiœdème se produit chaque semaine, et chez d’autres, seulement une ou deux fois par an. Une personne atteinte de cette maladie sur trois connaît au moins une fois dans sa vie une crise potentiellement mortelle.
Diagnostic
Le diagnostic peut être retardé de plusieurs années[28], ses symptômes ressemblant à ceux de maladies plus fréquentes, telles qu’une allergie ou une colique intestinale. L’échec de la réponse aux antihistaminiques ou aux corticoïdes livre une indication importante, en permettant une différenciation par rapport aux réactions allergiques. Le diagnostic est particulièrement difficile à poser chez les patients souffrant exclusivement de crises gastro-intestinales. Outre l’étude des antécédents familiaux, qui fournit la preuve de la maladie, seule une analyse en laboratoire peut en dernier ressort apporter des certitudes. En règle générale, ce n’est pas le déficit en C1-INH lui-même qui est établi, mais une baisse du taux du facteur du complément C4[29]. Celui-ci intervient dans la chaîne de réaction du système du complément, qui est constamment hyperactif en raison de l’absence de régulation par C1-INH. Le taux de C4 peut être cependant normal[30] et le diagnostic est alors affirmé par la mesure de la concentration de la C1-INH (type 1) ou de l'activité de cette dernière (type 2).
Un dosage de la C4 doit être fait chez les autres membres de la famille.
Le test génétique (recherche d'une mutation sur le gène SERPING1) n'est pas nécessaire pour établir le diagnostic. Plus de 400 mutations ont été identifiées[5].
Traitement
La prise en charge des angiœdèmes bradykiniques a fait l'objet de la publication de recommandations par plusieurs groupes d'experts internationaux en 2012[31] et en 2013 (recommandations américaines [32]).
Traitement de la crise
Il a pour objectif d’arrêter aussi vite que possible la progression ultérieure de l’œdème, ce qui peut sauver la vie du patient, notamment en cas de crise laryngée. Un concentré de C1-INH, extrait de sang de donneur, peut être administré par voie intraveineuse[33]. Il existe également une forme recombinante de la protéine.
L'icatibant est le premier antagoniste des récepteurs B2 de la bradykinine, principal médiateur de l'angiœdème bradykinique. En bloquant ces récepteurs, l'icatibant inhibe ce médiateur donne cependant des résultats mitigées dans le traitement d'attaque[34]. Le , l'Agence Européenne pour l'Évaluation des Médicaments a délivré une autorisation de mise sur le marché pour l'icatibant, premier traitement par voie sous-cutanée, disponible dans toute l'Europe pour les crises aiguës de l'angiœdème bradykinique. L'ecallantide[35](inhibieur de la kallikréine) et le lanadelumab[36] (un anticorps monoclonal dirigé contre la kallicréine), semblent avoir une bonne efficacité sur la crise ou sur sa prévention. Le sebetralstat, en cours de test, est aussi un inhibiteur de la kallicréine mais qui peut être donné par voie orale, permettant une amélioration plus rapide des symptômes[37].
Prévention à long terme
À cette fin, on utilisait souvent des hormones sexuelles mâles (androgènes), (Berinert P, danazol[38]), par exemple), qui augmentent la production de C1-INH dans le foie par le biais d’un mécanisme jusqu’à présent non élucidé. La dose doit être aussi basse que possible, en raison de sérieux effets secondaires comme une prise de poids importante, de la dépression, un phénomène de virilisation chez les femmes, de l'hypertension et une augmentation du cholestérol (LDL). L’utilisation des androgènes est problématique notamment chez les enfants ; elle est contre-indiquée chez les femmes enceintes. L’apparition de tumeurs hépatiques bénignes, observée dans plusieurs cas sous traitement par l’androgène danazol, a entraîné le retrait du marché de cette substance au début de 2005 en Allemagne.
La perfusion de la protéine déficiente permet le raccourcissement des crises[39].
Chez les patients ayant fait une crise sous un inhibiteur de l'enzyme de conversion, l'arrêt de ce dernier est impératif. Il peut être mis alors sous un antagoniste des récepteurs de l'angiotensine II, mais cela n'annule pas complètement le risque de récidive[40].
À titre de remplacement, on utilise donc aussi les inhibiteurs de la fibrinolyse, tels que l’acide tranexamique[41], qui présentent toutefois des effets thérapeutiques relativement faibles et des effets secondaires gênants.
DX-88 est un inhibiteur de la kallikréine en développement comme médicament orphelin pour l’angiœdème bradykinique. L’icatibant est un antagoniste sélectif du récepteur de la bradikinine, qui doit être commercialisé à titre de médicament orphelin pour l’angiœdème bradykinique par Jerini AG, une entreprise pharmaceutique allemande[42], une société néerlandaise de biotechnologie développe un inhibiteur C1 recombinant pour les crises aiguës d’angiœdème bradykinique. Le produit rhC1INH de Pharming et l’icatibant de Jerini sont actuellement au stade III du développement et bénéficient du statut de médicament orphelin aux États-Unis et en Europe.
Une autre piste de recherche est l'utilisation d'un ARN anti sens permettant la destruction de l'ARN messager de la pré- kallikréine, le donidalorsen. Ce dernier permet ainsi la réduction du nombre de crises[43].
Prévention à court terme
La prévention à court terme est généralement utilisée avant des procédures chirurgicales ou un traitement dentaire. On administre à cette fin le concentré de C1-INH, 1 ou 1 ½ heure avant l’intervention, ce qui est possible par voie sous-cutanée[44]. Dans les pays où le concentré de C1 inhibiteur n’est pas disponible, ou dans lesquels ce concentré n’est disponible que pour les urgences (œdèmes laryngés) un traitement par fortes doses d’androgènes est administré pendant 5-7 jours.
Historique
La maladie, ainsi que son caractère héréditaire, a été décrite en 1888 par William Osler[45]. Le rôle du déficit en inhibiteur de la C1 estérase a été reconnu en 1963[12].
Le traitement par C1-INH a été décrit pour la première fois en 1976[46]. Extrait du sang humain, le risque viral a fait limiter sa prescription dans certains pays, notamment aux USA[47]. Le risque a été considérablement réduit, dans un premier temps par un produit pasteurisé, puis ultrafiltré.
Notes et références
- Orphanet
- « Angiœdème bradykinique », Orphanet Urgence, (lire en ligne [PDF])
- Hereditary Angioedema
- Stéphanie Petitpierre, Pierre-Alexandre Bart, François Spertini et Annette Leimgruber, « Multiples étiologies de l'angiœdème [The multiple etiologies of angioedema] », Rev. med. suisse, vol. 4, no 154, (PMID 18557532, lire en ligne, consulté le )
- Busse PJ, Christiansen SC, Hereditary angioedema, N Engl J Med, 2020;382:1136-1148
- (en) A. Bygum « Hereditary angio-oedema in Denmark: a nationwide survey » Br J Dermatol. 2009;161:1153-8.
- B de Wazières & al. ; L'œdème angio-neurotique héréditaire : une urgence médicale sous-estimée : à propos de 33 cas (de 1973 à 1990) = Hereditary angio-oedema:an underestimated threatening disease:a report of 33 cases ; Annales de dermatologie et de vénéréologie ; ISSN 0151-9638 ; 1995, vol. 122, no1-2, pp. 11-15 (27 ref), Résumé Inist/CNRS
- J.L. Dupond, Ph. Humbert, B. Hor, D. Wendling, B. de Wazières, Th. Fest, D. Vuitton (1992), L'œdème angio-neurotique héréditaire : une maladie congénitale sous estimée. À propos de 36 cas (XXVIIème Congrès de la SNFMI Société Nationale Française de Médecine Interne) ; La Revue de médecine interne ; Volume 13, Issue 7, December 1992, Pages S402 (Résumé)
- E. Delaporte, M. Géniaux, J.-P. Lacour, L. Vaillant (2002), Allergies cutanéo-muqueuses chez l’enfant et l’adulte Urticaire et œdème de Quincke ; Examen National Classant (Module transdisciplinaire 8 : Immunopathologie, réactions inflammatoires) ; Ann Dermatol Venereol ; 129:2S83-2S89
- P Bielefeld, M Guiguet, P Meyer, RO Casasnovas, D Caillot, JF Besancenot (1994), Œdème angioneurotique acquis et lymphome de bas grade : problèmes thérapeutiques. À propos de deux nouvelles observations ; La Revue de médecine interne, Volume 15, Supplement 1, 1994, Page 138s (Résumé)
- « Résumé des caractéristiques du produit - Plaquenil 200 mg, comprimé pelliculé - Base de données publique des médicaments », sur base-donnees-publique.medicaments.gouv.fr (consulté le )
- (en) Donaldson VH, Evans RR. « A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C' 1-esterase » Am. J. Med. 1963;35:37-44
- (en) Rosen FS, Pensky J, Donaldson V, Charache P. « Hereditary angioneurotic edema: two genetic variants » Science 1965;148:957-8.
- (en) Longhurst H, Cicardi M, Hereditary angio-oedema, Lancet, 2012;379:474-481
- (en) Pappalardo E, Caccia S, Suffritti C, Tordai A, Zingale LC, Cicardi M. « Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angio-oedema: functional and structural correlates » Mol Immunol. 2008;45:3536-44.
- HAEdb-C1 inhibitor gene mutation database. http://hae.enzim.hu/stat.php. 2008. 7-1-2010. (nach [19]).
- Agostoni A, Aygoren-Pursun E, Binkley KE, Blanch A, Bork K, Bouillet L, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004;114:S51–131.
- (en) Davis AE, « Hereditary angioedema: a current state-of-the-art review. III. Mechanisms of hereditary angioedema » Ann Allergy Asthma Immunol. 2008;100:S7-S12
- ONTARGET investigators, Yusuf S, Teo KK, Pogue J et al. « Telmisartan, ramipril, or both in patients at high risk for vascular events » N Engl J Med. 2008;358:1547
- (en) Cugno M, Nussberger J, Cicardi M, Agostoni A. « Bradykinin and the pathophysiology of angio-oedema » Int Immunopharmacol. 2003;3:311-7.
- (en) Bork K, Fischer B, Dewald G. « Recurrent episodes of skin angio-oedema and severe attacks of abdominal pain induced by oral contraceptives or hormone replacement therapy » Am J Med. 2003;114:294-8.
- (en) Farkas H, Füst G, Fekete B, Karádi I, Varga L. Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema Lancet 2001;358:1695-6.
- (en) Zanichelli A, Vacchini R, Badini M, Penna V, Cicardi M, Standard care impact on angio-oedema because of hereditary C1 inhibitor deficiency: a 21-month prospective 2 study in a cohort of 103 patients, Allergy, 2010;66:192-196
- Zotter Z, Csuka D, Szabó E et al. The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency, Orphanet J Rare Dis, 2014;9:44-44
- (en) Prematta MJ, Kemp JG, Gibbs JG, Mende C, Rhoads C, Craig TJ. « Frequency, timing, and type of prodromal symptoms associated with hereditary angio-oedema attacks » Allergy Asthma Proc. 2009;30:506-11.
- (en) Bork K, Hardt J, Schicketanz KH, Ressel N. « Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency » Arch Intern Med. 2003;163:1229-35.
- (en) Farkas H, Harmat G, Kaposi PN et al. « Ultrasonography in the diagnosis and monitoring of ascites in acute abdominal attacks of hereditary angioneurotic oedema » Eur J Gastroenterol Hepatol. 2001;13:1225-1230
- (en) Roche O, Blanch A, Caballero T, Sastre N, Callejo D, López-Trascasa M. Hereditary angio-oedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain Ann Allergy Asthma Immunol. 2005;94:498-503.
- (en) Gompels MM, Lock RJ, Morgan JE, Osborne J, Brown A, Virgo PF. « A multicentre evaluation of the diagnostic efficiency of serological investigations for C1 inhibitor deficiency » J Clin Pathol. 2002;55:145-7.
- (en) Tarzi MD, Hickey A, Förster T, Mohammadi M, Longhurst HJ. « An evaluation of tests used for the diagnosis and monitoring of C1 inhibitor deficiency: normal serum C4 does not exclude hereditary angio-oedema » Clin Exp Immunol. 2007;149:513-6.
- (en) Cicardi M, Bork K, Caballero T for the HAWK (Hereditary Angioedema International Working Group). « Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group » Allergy 2012;67:147-57.
- Zuraw BL, Banerji A, Bernstein JA et al. US Hereditary Angioedema Association Medical Advisory Board 2013 recommendations for the management of hereditary angioedema due to C1 inhibitor deficiency, J Allergy Clin Immunol Pract, 2013;1:458-467
- (en) Craig TJ, Levy RV, Wasserman RL, « Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks » J Allergy Clin Immunol. 2009;124:801-8.
- (en) Cicardi M, Banerji A, Bracho F et al. « Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema » N Eng J Med. 2010;363:532-541
- (en) Cicardi M, Levy RJ, McNeil DL et al. « Ecallantide for the treatment of acute attacks in hereditary angioedema » N Engl J Med. 2010;363:523-531
- Banerji A, Busse P, Shennak M et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis, N Engl J Med, 2017; 376:717-728
- Aygören-Pürsün E, anichelli A, Cohn DM et al. An investigational oral plasma kallikrein inhibitor for on-demand treatment of hereditary angioedema: a two-part, randomised, double-blind, placebo-controlled, crossover phase 2 trial, Lancet, 2023;401:458-469
- (en) Gelfand JA, Sherins RJ, Alling DW, Frank MM. « Treatment of hereditary angioedema with danazol: reversal of clinical and biochemical abnormalities » N Eng J Med. 1976;295:1444-8.
- Zuraw BL, Busse PJ, White M et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema, N Engl J Med, 2010;363:513-522
- Haymore BR, DeZee KJ, « Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor » Ann Allergy Asthma Immunol. 2009;103:83-4.
- (en) Frank MM, Sergent JS, Kane MA, Alling DW, Epsilon aminocaproic acid therapy of hereditary angioneurotic edema: a double-blind study, N Engl J Med, 1972;286:808-812
- Pharming
- Fijen LM, Reidl MA, Bordone L et al. Inhibition of prekallikrein for hereditary angioedema, N Engl J Med, 2022;386:1026-1033
- Longhurst H, Cicardi M, Craig T et al. Prevention of hereditary angioedema attacks with a subcutaneous C1 inhibitor, N Engl J Med, 2017;376:1131-1140
- Osler W, Hereditary angio-neurotic oedema, Am J Med Sci, 1888;95:362
- (it) Marasini B, Cicardi M, Agostoni A. « Trattamento dell'angioedema ereditario con concentrato plasmatico di inibitore della C1 esterasi [Treatment of hereditary angioedema with a plasmatic concentrate of CU esterase inhibitor] » Boll Soc Ital Cardiol. 1976;21:1449-52.
- (en) P. Morgan « Hereditary angioedema — Therapies old and new » N Engl J Med. 2010;363:581-3.
Sources
- Cet article est partiellement ou en totalité issu de l'article intitulé « Œdème de Quincke » (voir la liste des auteurs).
Lien externe
- Fiche Œdème angioneurotique sur Orpha.net