Amyotrophie spinale
L'amyotrophie spinale est le nom donné à un groupe de maladies héréditaires caractérisées par une faiblesse et une atrophie des muscles.
| Médicament | Nusinersen |
|---|---|
| Spécialité | Neurologie |
| OMIM | 253300 253550 253400 271150 |
|---|---|
| DiseasesDB | 14093 32911 12315 |
| MedlinePlus | 000996 |
| eMedicine | 1264401 |
| MeSH | D009134 |
| GeneReviews | Spinal Muscular Atrophy |
| Patient UK | Spinal-muscular-atrophy-pro |
![]() Mise en garde médicale
Mise en garde médicale
Cette pathologie se transmet de manière autosomique récessive.
Elle n'affecte pas les fonctions du cerveau mais peut conduire au décès des enfants en bas âge dans les cas les plus sévères. La raison principale est la mort des neurones moteurs conduisant à un non-fonctionnement des muscles, dont ceux nécessaires à la respiration. Les respirateurs artificiels sont souvent nécessaires, mais de nouveaux traitements existent (Thérapie antisens, thérapie génique…)
Symptômes
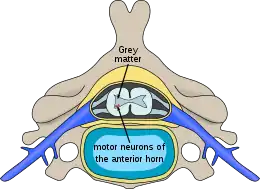
La maladie s'attaque et entraine la mort de certaines cellules nerveuses, les motoneurones qui stimulent et commandent les muscles volontaires. À cause de cela, les neurones moteurs qui se trouvent dans la corne antérieure de la moelle spinale ne sont plus en mesure de transmettre des signaux aux muscles, ce qui empêche ces derniers de fonctionner normalement. Par conséquent, les tissus musculaires s'affaiblissent puis s'atrophient (fondent). Cela n’affecte jamais les fonctions cognitives dites intellectuelles.
Il existe quatre types d'amyotrophie spinale qui présentent une apparition des symptômes selon l'âge et une sévérité différente. De manière générale, on peut remarquer une faiblesse des muscles et une faible tonicité musculaire.
Épidémiologie
L'amyotrophie spinale touche aussi bien les personnes de sexe féminin ou masculin. Près de 1 naissance sur 10 000 est concernée par cette maladie[1], ce qui représente environ 120 à 150 nouveau-nés, annuellement, en France[2].
La maladie est de type autosomique récessive. Si le père et la mère sont tous deux porteurs sains, l'enfant présente un risque sur quatre de développer une amyotrophie spinale[3]. L'allèle muté est présent chez environ une personne sur 54[1].
Cause
Sur le chromosome 5 sont situés deux gènes : SMN1 (ou SMNt - télomérique) et SMN2 (ou SMNc - centromérique). Ces deux gènes produisent la même protéine appelée SMN pour Survival Motor Neuron qui permet la survie des neurones moteurs. Ils diffèrent l'un de l'autre par la substitution de 5 acides aminés. La plus importante des différences se situe dans l'exon 7. Celle-ci rend 90 % de la protéine, synthétisée par le gène SMN2, tronquée donc non fonctionnelle.
Chez les patients, en l’absence de gène SMN1, la gravité de la maladie dépend du nombre de copies du gène SNM2, seuls responsables de la production de la protéine SMN[4].
Classification
Dénomination
Cette affection est décrite sous des appellations variées[5] :
- en français :
- AS (Amyotrophie Spinale)
- Atrophie musculaire progressive
- Amyotrophie spinale distale
- Amyotrophie spinale scapulopéronière
- Amyotrophie spinale de forme scapulo-péronière
- Syndrome de Stark-Kaeser
- Amyotrophie spinale oculopharyngée
- Amyotrophie spinale progressive
- Amyotrophie spinale proximale progressive
- Amyotrophie spinale progressive proximale
- Neuropathie bulbospinale
- Amyotrophie spinale de l'adulte
- Amyotrophie spinale de l'adulte type 4
- Amyotrophie spinale de l'adulte type IV
- en anglais :
- Spinal Muscular Atrophy
- Scapuloperoneal Form of Spinal Muscular Atrophy
- Oculopharyngeal Spinal Muscular Atrophy
- Progressive Muscular Atrophy
- Bulbospinal Neuronopathy
Classification
Parmi les amyotrophies spinales, on distingue quatre types (deux types infantiles, un type juvénile et un type de l’adulte)[3] :
- le type I, appelé amyotrophie spinale infantile sévère, apparaissant avant l’âge de 6 mois et caractérisé par l’absence d’acquisition de la station assise. Il existe des sous-types d’ASA de type I de sévérité variable ;
- le type II, ou amyotrophie spinale infantile intermédiaire, survenant à l’âge de 6 à 18 mois et caractérisé par l’absence d’acquisition de la marche ;
- le type III, aussi appelé amyotrophie spinale juvénile, survenant après l’âge d’acquisition de la marche (après 18 mois - 2 ans) ;
- le type IV ou amyotrophie spinale adulte, se manifestant à l’âge adulte. Il s’agit de diverses formes de la même maladie présentant des différences considérables du point de vue de l’âge d’apparition, de la gravité des symptômes et du pronostic. La mobilité peut être gravement altérée, mais l'espérance de vie n'est pas réduite.
Les trois premiers types sont regroupés sous le terme « amyotrophies spinales infantiles » (ASI), alors que le terme « amyotrophie spinale antérieure » (ASA) fait référence à l’ensemble des quatre types.
L'acronyme anglais SMA, pour spinal muscular atrophy, est également utilisé.
Le plus grand danger vient des affections respiratoires, pneumonie ou autres, contre lesquelles l'enfant combat difficilement.
Il existe des types alternatifs appelés « Bis » et « Tierce » ainsi que des classifications allant de la forme aiguë (maladie de Werdnig-Hoffman correspondant au type I) à la forme chronique (syndrome de Kugelberg-Welander correspondant au type III) en passant par des formes intermédiaires.
Les catégories évoquées là sont remises en cause car elles ne seraient ni objectives ni réalistes à cause des différences importantes de qualité de vie des patients (ex. : un type III peut être dans un état clinique plus grave qu'un type I bis)
Espérance de vie
L'espérance de vie varie énormément selon la forme d'amyotrophie spinale.
Avant l'ère des premiers traitements, voici ce que la littérature scientifique reportait :
- Type I : Espérance de vie moyenne courte d'environ 2 à 4 ans ;
- Type II : Espérance de vie moyenne d'environ 20 à 30 ans ;
- Type III : Espérance de vie normale ;
- Type IV : Espérance de vie normale.
Malgré l'amélioration de la prise en charge, notamment grâce à la ventilation artificielle, l'espérance de vie s'allonge mais reste encore à améliorer.
Les données long terme à la suite de l'initiation des nouveaux traitements permettront de revoir ces chiffres dans quelques années.
Traitement
En décembre 2016, le premier médicament pour l'amyotrophie spinale, une thérapie antisens (ARN) nommé nusinersen (Spinraza®), a été autorisé aux États-Unis par la FDA puis rapidement en Europe par l'EMA[6] à la suite de résultats significatifs sur la survie de nourissons atteints d'amyotrophie spinale de type I[7]. Cet ARN (Acide RiboNucléique ou oligonucléotide antisens) permet l'augmentation de l'expression de la protéine SMN normale issue du gène SNM2 en empêchant l'exclusion de l'exon 7. Après son initiation, ce traitement nécessite 3 injections annuelles par ponction lombaire pour atteindre directement la moelle spinale (là où se situent les motoneurones). Aujourd'hui, les résultats se confirment également chez les patients adultes atteints d'amyotrophie spinale[8].
En 2018, alors que des experts préfèreraient obtenir plus de données cliniques pour vérifier la fiabilité et l'innocuité du traitement sur du long terme, des groupes de familles et certains membres du Congrès poussent le Département de la Santé et des services sociaux, et son administrateur (Alex Azar), à ajouter dès 2018 cette maladie au panel de tests génétiques recommandés pour tous les nouveau-nés (dépistage néonatal)[9]. En 2019, cette initiative a été renforcée par la publication de premiers résultats du traitement chez des nourrissons présymptomatiques[10]. Enfin en aux États-Unis, 18 États avaient inclus l'amyotrophie spinale dans leur dispositif de dépistage néonatal[11].
De nouvelles études cliniques sont également en cours, utilisant des molécules chimiques ou la thérapie génique[12].
Dans le cas de la thérapie génique, le médicament consiste à injecter par voie intraveineuse un virus (virus adéno-associé) porteur du gène SMN1 afin qu'il exprime la protéine SMN manquante dans les cellules pour permettre l'amélioration des symptômes et de la survie des formes juvénile de type 1[13]. En , l'onasemnogene abeparvovec (en) (Zolgensma), dont l'action repose sur les principes cités ci-dessus a reçu l'autorisation de mise sur le marché aux États-Unis[14].
Il est à noter que le prix de ces médicaments, revendiqué par les laboratoires qui les ont mis au point, est très élevé et déconnecté du coût réel de la recherche[15] - [16].
Dépistage
En France en 2023, le programme Depisma évalue le dépistage néonatal universel dans deux régions : le Grand Est et la Nouvelle-Aquitaine[17] où l'incidence est d'un bébé sur 7000[18]. Le dépistage s'effectue par prélèvement sanguin « au buvard ». Il a pour objet la détection de la mutation homozygote du gène SMN1. L'intérêt de ce dépistage chez le nourrisson présymptomatique est mis en évidence par une étude publiée en janvier 2023[19].
Notes et références
- Sugarman EA, Nagan N, Zhu H et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens, Eur J Hum Genet, 2012;20:27-32
- J. Andoni Urtizberea et Ferroudja Daidj, « Combien de patients atteints de SMA en France ? », médecine/sciences, vol. 34, , p. 32–34 (ISSN 0767-0974 et 1958-5381, DOI 10.1051/medsci/201834s209, lire en ligne, consulté le )
- (en) Adele D'Amico, Eugenio Mercuri, Francesco D. Tiziano, Enrico Bertini, « Spinal muscular atrophy », Orphanet Journal of Rare Diseases, vol. 6, no 1, , p. 71 (ISSN 1750-1172, PMID 22047105, DOI 10.1186/1750-1172-6-71, lire en ligne, consulté le )
- Mailman MD, Heinz JW, Papp AC et al., Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2, Genet Med, 2002;4:20-26
- « Descripteur français : Amyotrophie spinale », sur mesh.inserm.fr
- (en) Anonymous, « Spinraza », sur European Medicines Agency, (consulté le )
- Richard S. Finkel, Eugenio Mercuri, Basil T. Darras et Anne M. Connolly, « Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy », New England Journal of Medicine, vol. 377, no 18, , p. 1723–1732 (ISSN 0028-4793, PMID 29091570, DOI 10.1056/NEJMoa1702752, lire en ligne, consulté le )
- (en) Tim Hagenacker, Claudia D. Wurster, René Günther et Olivia Schreiber-Katz, « Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study », The Lancet Neurology, vol. 19, no 4, , p. 317–325 (ISSN 1474-4422 et 1474-4465, PMID 32199097, DOI 10.1016/S1474-4422(20)30037-5, lire en ligne, consulté le )
- (en) Wadman M (2018) « Newborn screening urged for fatal neurological disorder » ; Science mag 29 Juin : vol. 360, (no), p. 1385 DOI 10.1126/science.360.6396.1385
- (en) Darryl C. De Vivo, Enrico Bertini, Kathryn J. Swoboda et Wuh-Liang Hwu, « Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study », Neuromuscular Disorders, vol. 29, no 11, , p. 842–856 (ISSN 0960-8966 et 1873-2364, PMID 31704158, DOI 10.1016/j.nmd.2019.09.007, lire en ligne, consulté le )
- (en-US) Grace Frank, « 18 States in US Screening Newborns for SMA as Efforts Continue for... », sur SMA News Today, (consulté le )
- (en) Karen Weintraub, « A boy’s unlikely survival raises the curtain on a new class of drugs », MIT Technology Review, (lire en ligne, consulté le )
- (en) J. R. Mendell, S. Al-Zaidy et R. Shell et al., « Single-dose gene-replacement therapy for spinal muscular atrophy », N. Engl. J. Med., no 377, , p. 1713-1722 (lire en ligne)
- « Une première historique : une thérapie génique autorisée aux États-Unis pour une maladie neuromusculaire », sur https://www.afm-telethon.fr, (consulté le )
- « L’OMS appelle à plus de transparence sur le prix des médicaments ! L’AFM-Téléthon souhaite des mesures concrètes », sur https://www.afm-telethon.fr, (consulté le )
- Isabelle Barré, « Le casse d'un géant pharmaceutique sur une merveille de la recherche française », Le Canard enchaîné, no 5144, , p. 4
- Damien Coulomb, « Amyotrophie spinale : le dépistage néonatal améliore la capacité des enfants à marcher », sur Le Quotidien du médecin, .
- Isabelle Castéra, « Amyotrophie spinale : tous les bébés néo-aquitains seront désormais testés », sur Sud Ouest, .
- (en) Didu S Kariyawasam, Arlene M D'Silva, Hugo Sampaio, Nancy Briggs, Karen Herbert, Veronica Wiley, Michelle A Farrar, « Newborn screening for spinal muscular atrophy in Australia: a non-randomised cohort study », The Lancet, Elsevier, (ISSN 0140-6736 et 1474-547X, OCLC 01755507, DOI 10.1016/S2352-4642(22)00342-X)
Voir aussi
- Maladie de Werdnig-Hoffmann ou amyotrophie spinale de type I
- Syndrome de Kugelberg-Welander ou amyotrophie spinale de type III