Synthèse peptidique
En chimie organique, la synthèse peptidique[1] - [2] est la production de peptides, des composés organiques, dans lesquels des acides aminés sont liés par l'intermédiaire de liaisons amide, qui dans ce cas prennent le nom de liaisons peptidiques. Le processus biologique de la production de peptides longs (protéines) est connu comme la biosynthèse des protéines.
Chimie
Les peptides sont synthétisés par le couplage du groupe carboxyle d'un acide aminé avec le groupe amino de l'acide aminé suivant dans la molécule. En raison du risque d'induire des réactions parasites non-désirées, la protection préalable des groupes fonctionnels est généralement nécessaire. La synthèse chimique des peptides démarre en général par l'extrémité carboxyle et se déroule en direction de l'extrémité amino-terminale, c'est-à-dire en sens inverse de la synthèse biologique des protéines.
Synthèse en phase liquide
La synthèse peptidique en phase liquide est une méthode classique. Dans la plupart des applications pratiques, elle a cependant été remplacée par la synthèse sur support solide. Elle conserve cependant une utilité importante pour les applications de production de peptide à grande échelle à des fins industrielles.
Synthèse sur support solide
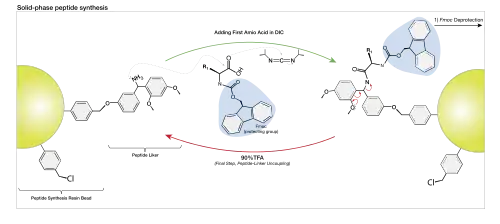
La synthèse peptidique sur support solide (SPPS, pour solid phase peptide synthesis) mise au point par Robert Merrifield[3] est devenue la méthode de référence pour la synthèse de peptides et de protéines au laboratoire. La SPPS permet la synthèse de peptides naturels difficiles à produire dans des bactéries, l'incorporation d'acides aminés non-naturels, ou d'effectuer des modifications du squelette peptidique de peptides et de protéines.
Des billes de résine poreuses sont greffées avec des liens espaceurs (les "linkers") sur lesquels les chaînes peptidiques peuvent ensuite être construites. Le peptide reste fixé de manière covalente à la bille de résine jusqu'à son clivage final par un réactif tel que le fluorure d'hydrogène anhydre ou l'acide trifluoroacétique. Le peptide est ainsi "immobilisé" sur le support solide et reste accroché lors de rinçages destinés à éliminer les réactifs et sous-produits de synthèse restés dans la phase liquide.
Le principe général de la SPPS consiste à répéter des cycles de déprotection, de lavage, de couplage et de lavage. L'amine terminale du peptide immobilisée est couplée à un acide aminé dont l'extrémité N-terminale est elle bloquée par un groupement protecteur (voir ci-dessous). Ce nouvel acide aminé est ensuite déprotégé, révélant une nouvelle amine N-terminale de l'amine à laquelle un autre acide aminé peut ensuite être attaché. La supériorité de cette technique réside en partie dans la possibilité d'effectuer des cycles de lavage après chaque réaction, en éliminant les excès de réactif tandis que le peptide d'intérêt reste fixé de manière covalente à la résine solide.
Il existe deux stratégies de synthèse sur support solide, la chimie Fmoc et la chimie Boc, acronymes des groupes protecteurs utilisés pour bloquer les groupements amine. Contrairement à la synthèse protéique par les ribosomes, la synthèse peptidique sur support solide s'effectue du C-terminal vers le N-terminal. Il existe des synthétiseurs automatiques qui permettent de mettre en œuvre ces deux techniques, même si la synthèse manuelle est parfois encore utilisée.
Groupes protecteurs
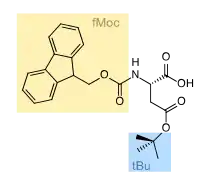
La performance de la synthèse peptidique repose sur la capacité à éliminer les réactions parasites qui font chuter le rendement lors des cycles répétitifs de synthèse. Dans la synthèse SPPS en phase solide, le groupement acide carboxylique terminal est protégé par le lien avec le support. Il reste donc à protéger la fonction α-amine du squelette peptidique et les groupements réactifs éventuellement présents sur la chaîne latérale, en fonction de la nature de l'acide aminé à coupler à chaque cycle.
Les stratégies de protection des chaînes latérales et de l'amine du squelette doivent être orthogonales, c'est-à-dire que la protection des chaînes latérales doit être stable lors des cycles de déprotection de la fonction α-amine. Les chaînes latérales ne sont en effet déprotégées qu'en toute fin de synthèse, pour éviter des réactions parasites qui produiraient des peptides ramifiés.
Groupements protecteurs des amines
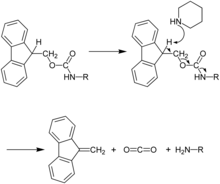
Il y a deux grands types de groupements protecteurs des amines utilisés en synthèse peptidique : le tert-butoxycarbonyle ou Boc et le fluorénylméthoxycarbonyle ou Fmoc. Pour la déprotection, le Boc s'élimine par un traitement acide (acide trifluoroacétique) et le Fmoc par un traitement basique (pipéridine).
Il est possible de protéger conjointement les fonctions α-amine par des groupes Fmoc et les amines des chaînes latérales (lysine) par des groupes Boc, la déprotection du Fmoc à la pipéridine étant sans effet sur les Boc qui restent fixés lors des cycles répétitifs de couplage des acides aminés.
Le groupement carboxybenzyle (ou Cbz ou Z) est également utilisé, essentiellement pour la protection orthogonale des amines de chaînes latérales dans la stratégie de synthèse Boc. La déprotection s'effectue par une hydrogénation catalytique à la fin de la synthèse.
Couplage
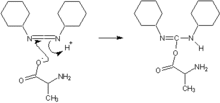
Après déprotection de l'amine N-terminale du peptide en cours de synthèse, on ajoute le synthon correspondant à l'acide aminé suivant dans la séquence. Pour que la liaison peptidique se forme entre le groupement carboxyle du synthon et l'amine du peptide, il est nécessaire d'ajouter un agent de couplage pour activer le carboxyle.
On utilise classiquement deux grands types d'agents de couplage : les carbodiimides, parfois en conjonction avec les triazolols[4].
Le dicyclohexylcarbodiimide, le plus employé des carbodiimides, forme un intermédiaire réactif O-acylisourée avec le carboxyle, qui subit ensuite une attaque nucléophile par l'amine libre du peptide.
Les triazolols comme le 1-hydroxy-benzotriazole (HOBt) stabilisent l'acylisourée intermédiaire et limitent le risque de racémisation de l'acide aminé.
Articles connexes
Références
- Stephen B. H. Kent, « Chemical Synthesis of Peptides and Proteins », Annual Review of Biochemistry, vol. 57, no 1, , p. 957–989 (ISSN 0066-4154, DOI 10.1146/annurev.bi.57.070188.004521, lire en ligne, consulté le )
- (en) Miklos Bodzansky, Principles of Peptide Synthesis, Springer, , 329 p. (ISBN 978-3-642-78056-1, lire en ligne)
- R. B. Merrifield, « Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide », J. Am. Chem. Soc., vol. 85, no 14, , p. 2149–2154 (DOI 10.1021/ja00897a025)
- (en) Fernando Albericio, Solid-Phase Synthesis. A practical guide, CRC Press, , 848 p. (ISBN 978-0-8247-0359-2, lire en ligne)