Sclérodermie systémique
La sclérodermie systémique (ScS) ou sclérodermie systémique progressive ou « sclérose systémique », est l'une des diverses formes de sclérodermies. C'est une affection disséminée du tissu conjonctif, entrant dans le groupe des connectivites. Elle est caractérisée par une fibrose cutanée et vasculaire pouvant conduire à une défaillances d'organes vitaux (cœur notamment). Il s'agit d'une maladie rare qui fait partie des maladies orphelines.
| Médicament | Chloroquine, tolazoline (en), aminobenzoate potassium (d), ambrisentan et bosentan monohydraté (d) |
|---|---|
| Spécialité | Rhumatologie |
| CIM-10 | M34.0 |
|---|---|
| CIM-9 | 710.1 |
| OMIM | 181750 |
| DiseasesDB | 12845 |
| MedlinePlus | 000429 |
| eMedicine | 1066280 |
| MeSH | D012595 |
![]() Mise en garde médicale
Mise en garde médicale
Son origine est mal comprise, mais la maladie semble multifactorielle et avoir des causes au moins en partie environnementales, et notamment via certaines formes de la pollution de l'air (ex : + 25% de risque de développer la maladie chez les hommes exposés à l'inhalation de silice (particulaire[1] - [2] - [3]).
Il est également possible que la sclérodermie systémique soit liée à une infection chronique des cellules endothéliales et à un changement d'avidité pour l'oxygène des globules rouges lors d'hémobartonelloses chroniques, au vu de la superposition importante des symptômes entre formes chroniques d'hemobartonellose et sclérodermie systémique sine scleroderma.
Épidémiologie
Il s'agit d'une maladie relativement rare, dont la prévalence varie entre 7 et 500 cas par million d'habitants, avec une nette prédominance féminine[4]. Selon certains auteurs, cette maladie est retrouvée plus fréquemment chez les personnes à peau noire, mais alors avec des caractéristiques immunologiques un peu différentes[5].
La prévalence, mal connue dans les pays en développement, est « estimée entre 3 et 24 cas/100 000 habitants » dans les pays dits développés[1].
Causes et les mécanismes de la maladie
Les causes de ces maladies restent mal comprises.
Les déterminants familiaux et auto-immuns ne sont pas convaincants[1].
L'exposition de l'organisme à des toxiques semble pouvoir aggraver (potentialisation) la maladie (son extension sur la peau, son degré d'« atteinte pulmonaire infiltrante » ou encore son « profil immunologique »...)[1]. Des facteurs environnementaux semblent souvent en cause[6], professionnels[6] et occupationnels[1] - [7]. C'est ce que suggère l'existence de foyers sporadiques de plus forte prévalence. Les facteurs de risque avérés ou suspectés sont notamment :
- l'exposition à la silice ; ce facteur de risque, suggéré dès 1917, est actuellement reconnu au tableau 25 bis des maladies professionnelles[1] - [2] - [8] - [9] - [10]. Selon une étude publiée en 1990, les hommes exposés à la silice voient leur risque de développer une sclérodermie augmenter de 25 fois (par rapport aux hommes non-exposés)[11] ;
Le syndrome d'Erasmus est le nom qui a été donné à « l'association d'une sclérodermie générale progressive et d'une exposition à la silice avec ou sans silicose »[12] ; - les implants de silicone et de paraffine (implant mammaire notamment, et exposition externe au silicone[6] ; ce sont des facteurs de risque suspectés en raison de leur teneur en silice ;
- l'exposition au chlorure de vinyle (le composé de base du PVC) ; c'est une source démontrée de syndrome de Raynaud[13] ;
- l'ingestion de certaines huiles frelatées (syndrome de l'huile toxique[6]) ;
- la prise de L-tryptophane adultéré[6] ; un syndrome de type myalgies-éosinophilie a été induit par l'ingestion de L-tryptophane produit en réacteur par des bactéries issues du génie génétique. Le produit était probablement dénaturé par un contaminant non identifié. La maladie a disparu avec le retrait du produit du marché ;
- certaines intoxications médicamenteuses[2], par exemple par bléomycine[6], pentazocine[6] ;
- l'exposition à certains solvants, « incriminés dans plusieurs études cas-témoins de méthodologie rigoureuse leur conférant une imputabilité forte »[1] ;
- l'exposition à divers polluants ou autres toxiques ; les résines époxy et fumées de soudage sont suspectées, mais leur responsabilité dans la survenue ou la facilitation d'une sclérodermie systémique n'est pas encore démontrée[1] ;
- l'exposition à des vibrations, suspectée mais non démontrée[1].
Physiopathologie
La lésion initiale est une lésion des petits vaisseaux (artériole) avec anomalies de la couche cellulaire interne (endothelium) entraînant une extravasation de cellules inflammatoires et de liquide autour du vaisseau[14]. Elle conduit à une fibrose responsable des signes de la maladie[15].
Données cliniques
Le phénomène de Raynaud
Un syndrome de Raynaud s'observe dans 90 % des cas et le plus souvent il s'agit du symptôme inaugural. On estime que 5 à 10 % des patients présentant un syndrome de Raynaud complet, apparemment isolé, développeront une connectivite, le plus souvent une sclérodermie.
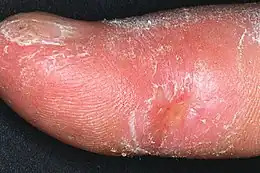
L'atteinte cutanée
L'atteinte si caractéristique de la peau est l'élément-clef du diagnostic.
- La sclérodactylie le plus souvent associée au syndrome de Raynaud, aboutit à des doigts effilés, durs et raides, sans aucune élasticité. L'atteinte gagne de proche en proche la main et le poignet. La sclérodactylie avec syndrome de Raynaud peut s'associer à des calcifications sous cutanées, des télangectasies et une atteinte œsophagienne pour former le Syndrome de CREST.
- L'atteinte du visage commence autour de la bouche et au front, effaçant les rides et donnant un aspect de peau cartonnée, voire marmoréenne.
- Dans la sclérodermie diffuse, l'infiltration atteint la racine des membres et le tronc. Cette forme est plus tardive, le pronostic en est moins bon. La peau est luisante, impossible à pincer, et le patient peut se trouver enserré dans une véritable cuirasse.
Le système locomoteur
- Atteintes ostéo-articulaires : Polyarthrites inflammatoires, tendinites, ostéolyse des extrémités,
- musculaires : Faiblesse musculaire des ceintures, pouvant s'accompagner d'anomalies enzymatiques et électriques d'atteinte myogène,
- neurologiques : Mononévrites, du trijumeau ou du canal carpien, voire polyneuropathies.
Digestives
L'atteinte œsophagienne concerne 75 % des patients, à plus ou moins longue échéance. Une paralysie progressive de la partie inférieure de l'œsophage provoque une dysphagie. Il existe un reflux provoquant une œsophagite parfois ulcérée. Il peut y avoir plus rarement des atteintes du même type au niveau de l'estomac et de l'intestin grêle, pouvant entrainer un syndrome de malabsorption. On a aussi décrit des télangiectasies digestives, sources d'hémorragies.
Pulmonaires
La sclérodermie peut atteindre les poumons avec développement d'une pneumopathie interstitielle diffuse pouvant évoluer vers une fibrose pulmonaire, plus ou moins fréquemment selon les séries publiées et le mode de diagnostic. Il peut aussi exister une hypertension artérielle pulmonaire, isolée ou non.
Cardiaques
Leur fréquence est très variable selon le type de séries publiées, cliniques, échocardiographiques ou anatomiques. Il n'y a pas d'atteinte endocardique. Les manifestations péricardiques sont classiques, mais c'est surtout l'atteinte myocardique qui est fréquente et sévère. À noter la possibilité de spasmes coronariens, véritables « Raynaud coronaires ».
Rénales
La « crise rénale sclérodermique » réalise une urgence vitale avec insuffisance rénale aiguë et hémolyse. La plupart du temps il s'agit cependant d'une insuffisance rénale chronique lentement progressive avec hypertension artérielle.
Les troubles auditifs
Les troubles auditifs ne sont pas rares, avec une prévalence de 50 % d'acouphènes, 40 % d'hyperacousie et 40 % de pertes auditives[16].
Les examens complémentaires
Il existe souvent dans la sclérodermie systémique un syndrome inflammatoire biologique rarement intense. Les anticorps antinucléaires sont positifs avec présence d'anticorps anti-centromères, d'anti-ADN topoisomérase I (anciennement les anti-Scl 70)
La capillaroscopie est un élément important du diagnostic précoce. La biopsie cutanée n'est pas nécessaire au diagnostic, et même déconseillée par certaines équipes du fait de possibles difficultés de cicatrisation.
Les profils immunologiques de la sclérodermie systémique
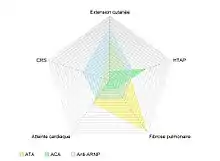
La relation entre les atteintes viscérales et les auto-anticorps retrouvés au cours de la sclérodermie systémique, notamment les anticorps anti-centromères, les anticorps anti-topoisomérase I et les anti-ARN polymérases, est largement rapportée dans la littérature[17]. Ces associations sont tellement étroites qu'il a été possible d'identifier 3 sous-groupes distincts par leurs manifestations cliniques, leurs profils sérologiques(spécificité mise en évidence) et leurs pronostics.
Évolution et pronostic
Le pronostic de la sclérodermie systémique est variable, fonction des complications, notamment pulmonaires, rénales et digestives. La maladie est à ce jour incurable, mais divers médicaments peuvent limiter une partie de ses effets néfastes sur l'organisme[18].
Formes « frontières »
- Forme œdémateuse : la maladie peut commencer par des œdèmes longtemps isolés qui font errer le diagnostic. Il ne faut pas confondre la sclérodermie systémique avec le syndrome de Shulman (ou fasciite à éosinophiles).
- Syndrome de CREST
- Syndrome de Sharp
Formes associées
La sclérodermie systémique peut être associée à un grand nombre de maladies, dont les plus fréquentes sont :
- le syndrome de Goujerot-Sjögren ;
- la thyroïdite de Hashimoto ;
- la cirrhose biliaire primitive ;
- le cancer du poumon (pour lequel il est difficile d'éliminer l'association « fortuite ») ;
- le cancer du sein.
Traitement
La plupart des traitements de fond se sont avérés décevants. On peut citer la corticothérapie, la D-pénicillamine, le facteur XIII, l'interféron.
Il semblerait qu'un traitement prolongé par les immunosuppresseurs classiques puisse s'avérer utile pour ralentir l'évolution de la maladie.
Les malades peuvent également bénéficier de traitements symptomatiques qui sont susceptibles d'être adaptés à chaque cas, en fonction des atteintes viscérales et de leurs complications.
Notes et références
- O Hallé, T Schaeverbeke, B Bannwarth, J Dehais" (1997), « Les facteurs d'environnement et les éléments iatrogènes dans la sclérodermie systémique et les syndromes apparentés. Revue de la littérature » (Environmental and iatrogenic factors in systemic sclerosis and related conditions: review of the literature) ; La Revue de médecine interne ; Volume 18, Issue 3, March 1997, Pages 219–229(résumé)
- De la Espriella, J., & Crickx, B. (1991). « Sclérodermies et états sclérodermiformes induits par la silice et des agents chimiques ou médicamenteux », Annales de dermatologie et de vénéréologie vol. 118, (no) 12, p. 948-953. Masson.
- Dangers, expositions et risques relatifs à la silice cristalline Rapport d’expertise collective - Saisine « n° 2015-SA-0236 - Silice cristalline » | mars
- Chifflot H, Fautzi B, Sordet C, Chatelus E, Sibilia J, Incidence and prevalence of systemic sclerosis: a systematic literature review, Semin Arthritis Rheum, 2008;37:223-235
- Reveille JD, Ethnicity and race in systemic sclerosis: how it affects susceptibility, severity, antibody genetics, and clinical manifestations, Curr Rheumatol Rep, 2003;5:160-167
- Crépy, M. N., & Conso, F. (1994). Sclérodermie et facteurs professionnels (ou environnementaux). Archives des maladies professionnelles et de médecine du travail, 55(2), 111-118 (résumé)
- Nietert PJ, Silver RM, Systemic sclerosis: environmental and occupational risk factors, Curr Opin Rheumatol, 2000;12:520-526
- Koeger, A. C. (1994). « Responsabilité de l'exposition à la silice dans les connectivites », La Presse Médicale, 23(1), 11-14.
- Orlowski, E., De Cremoux, H., Pairon, J. C., Merlet, C., Bignon, J., & Brochard, P. (1990), « Exposition à la silice et maladies systémiques », Archives des maladies professionnelles de médecine du travail et de sécurité sociale, 51(8), 591-593.
- J.-B. Gaultier A. Hot P. Cathébras C. Grange J. Ninet H. Rousset (2008), « Sclérodermie systémique chez l’homme », La Revue de médecine interne 2008|29|3, p.181-186 (résumé).
- Granel, B., Zemour, F., Lehucher-Michel, M. P., Moulin, P., Disdier, P., Durand, J. M., ... & Frances, Y. (2008), « Évaluation de l’exposition toxique professionnelle de patients atteints de sclérodermie systémique. Revue de la littérature et résultat d’un auto-questionnaire ». La Revue de médecine interne, 29(11), 891-900. (résumé)
- Cointrel, C., Tillie-Leblond, I., Lamblin, C., Furon, D., TONNEL, A. B., & Wallaert, B. (1997). « Syndrome d'Erasmus : caractéristiques cliniques, tomodensitométriques, fonctionnelles respiratoires et biologiques du lavage bronchoalvéolaire », Revue des maladies respiratoires, 14(1), 21-26. (résumé avec Inist/CNRS))
- Infos (Informations médicales), Syndrome de Raynaud et sclérodermie (Immunologie), consulté 2013-10-06
- Prescott RJ, Freemont AJ, Jones CJ, Hoyland J, Fielding P, Sequential dermal microvascular and perivascular changes in the development of scleroderma, J Pathol, 1992;166:255-263
- Gabrielli A, Avvedimento EV, Krieg T, Scleroderma, N Eng J Med, 2009;360:1989-2003
- (en) Maciaszczyk K, Waszczykowska E, Pajor A, Bartkowiak-Dziankowska B, Durko T, « Hearing organ disorders in patients with systemic sclerosis » Rheumatol Int. 2011 ;31(11):1423-8. DOI 10.1007/s00296-010-1503-5
- M Koenig et M -J Fritzler, « Auto-anticorps et Sclérodermie Systémique: fréquence, associations cliniques, valeur pronostique et influence sur la progression des anomalies capillaroscopiques chez 307 patients canadiens-français », sur www.em-consulte.com, (consulté le )
- Laure Gossec, Jérôme Avouac, André Kahan, Yannick Allanore (2010), Sclérodermie systémique : critères diagnostiques et de suivi Systemic sclerosis: Diagnostic and follow-up criteria ; Critères et indices en rhumatologie : 2e partie ; Revue du Rhumatisme Monographies Volume 77, Issue 2, avril 2010, Pages 103–107 (résumé)