Maladie périodique
La maladie périodique (fièvre méditerranéenne familiale - (FMF), ou encore maladie arménienne) désigne une maladie génétique autosomique récessive qui se manifeste notamment par des poussées inflammatoires survenant à intervalles variables.
| Symptômes | Inflammation, fièvre, douleur abdominale, exanthème, douleur thoracique et myalgie |
|---|
| Médicament | (+/-)-Colchicine (d) |
|---|---|
| Spécialité | Rhumatologie |
| CIM-10 | E85.0 |
|---|---|
| CIM-9 | 277.31 |
| OMIM | 249100 608107 |
| DiseasesDB | 9836 |
| MedlinePlus | 000363 |
| eMedicine | 330284 |
| MeSH | D010505 |
| GeneReviews | |
| Patient UK | Familial-Mediterranean-Fever-(FMF)-Recurrent-Polyserositis |
![]() Mise en garde médicale
Mise en garde médicale
C'est la première maladie découverte (en 1945) et le chef de file du groupe des maladies auto-inflammatoires. Sa fréquence est la plus élevée chez les sujets originaires du pourtour méditerranéen (surtout juifs séfarades, sépharades, turcs et arméniens).
La maladie se manifeste dès l'enfance avec des accès fébriles et douloureux de périodicité variable. Sur le long terme, elle peut évoluer vers une insuffisance rénale grave par amylose. Un traitement continu par colchicine est efficace, dans plus de 90 % des cas, dans la prévention des accès inflammatoires et la survenue d'une amylose.
Histoire
La première observation de maladie périodique date de 1908 aux États-Unis : il s'agissait d'une jeune fille juive de 16 ans présentant une fièvre intermittente avec douleurs abdominales. En 1945, une description partielle de la maladie, réalisée à partir de 10 cas, est faite à New York[1]. La maladie est alors décrite comme finalement bénigne, malgré des accès aigus.
Elle est nommée « maladie périodique » en 1951 par Hobart Reimann (en), qui insiste sur son caractère cyclique[2]. L'année suivante les auteurs français Henry Mamou et Roger Cattan[3] en donnent la première description complète, en mentionnant son caractère familial et la gravité de l'atteinte rénale (amylose rénale)[4].
En 1956, Henry Mamou propose le terme épanalepsie (du grec ἐπανάληψις [epanálêpsis] qui signifie répétition)[5] dans son ouvrage La maladie périodique[6], mais le terme épanalepsie méditerranéenne restera inusité et finalement déconseillé[7]. De nombreux synonymes ont été proposés dont maladie des arméniens, polysérite récidivante ou familiale paroxystique, syndrome ou maladie de Siegal-Cattan-Mamou, etc.[7] - [8]
En anglais, elle est appelée « Familial Mediterranean Fever » en 1958, à cause de sa plus grande fréquence dans le bassin méditerranéen[1].
À partir de 1972, le rôle favorable de la colchicine comme traitement de fond est reconnu[9] - [10] - [11], ce qui transforme le pronostic de la maladie.
Dans les années 1980, des auteurs israéliens complètent l'étude clinique de la maladie et précisent son mode de transmission héréditaire et l'atteinte préférentielle de certaines ethnies[4].
En 1992, le gène de la maladie est localisé sur le bras court du chromosome 16[12]. En 1997, deux groupes de recherches identifient simultanément et séparément le gène de la maladie (identification de sa séquence génétique)[13].
Épidémiologie
La maladie périodique touche les populations du pourtour méditerranéen, principalement les turcs, les arméniens, les juifs séfarades et les juifs sépharades. Dans ces populations, la fréquence des porteurs du gène muté (porteurs sains) est de l'ordre de 1/7, de 1/3 à 1/20[14]. Cette prévalence élevée explique une transmission pseudo-dominante dans ces populations[15]. En Turquie, la prévalence de la maladie serait entre 1/150 à 1/10 000 ; chez les arméniens 1/1500 ; chez les juifs sépharades entre 1/250 et 1/1000[16] et chez les juifs séfarades entre 1/250 et 1/1000[16]. La maladie est aussi présente chez les juifs askhénazes 1/73 000, chez les grecs, les chypriotes, les italiens, les libanais, les druzes et les kurdes[16]. La non-appartenance à ces populations n'est pas un critère d'exclusion du diagnostic[14], des cas pouvant être signalés au Brésil, au Japon, ou en Europe de l'est[16].
Génétique

Le gène en cause est le gène MEFV (pour MEditerranean FeVer)[17] qui code une protéine, dite marénostrine (du nom latin de la méditerranée mare nostrum) ou pyrine (du grec feu, fièvre pyros), impliquée dans les processus inflammatoires[18] (activation de l'inflammasome)[1].
Ce gène a été identifié en 1997 par un consortium français[19]. Ce dernier a bénéficié d'une collaboration entre le programme Généthon et des partenaires turcs et tunisiens et de la participation d'un grand nombre de familles atteintes dans ces pays. Différentes mutations de ce gène apparaissent en effet chez 85 % des patients provenant de populations méditerranéennes. En parallèle, un autre collectif de chercheurs israéliens, américains et australiens identifiait également le lien entre ce gène et la maladie[20].
Une mutation à l'origine de cette maladie semble dater d'un peu plus de 2 000 ans, comme l'atteste la présence d'une variété du gène mutant chez des populations juives irakiennes restées isolées des autres populations juives depuis 2 500 ans.
Ce gène est exprimé dans les granulocytes, les monocytes et les éosinophiles : ces leucocytes sont impliqués dans les mécanismes inflammatoires. Il reste maintenant à établir le mécanisme d'action de la protéine découverte et de déterminer comment agit la colchicine.
Une piste ouverte est celle de l'étude de l'interaction entre la protéine codée par le gène et les mécanismes inflammatoires : si un lien est avéré, de nouvelles molécules anti-inflammatoires pourraient éventuellement être développées.
Physiopathologie
La physiopathologie de la fièvre méditerranéenne familiale a connu récemment des avancées importantes: la pyrine est un récepteur qui peut former un inflammasome[21]. A l'état basal, elle est maintenue inactive par une protéine chaperonne (de la famille des protéines 14.3.3) liée à la pyrine au niveau de résidus sérines phosphorylés[22]. La déphosphoration de ces sérines (localisées dans le linker codé par l'exon 2 de MEFV) est un préalable indispensable à l'activation de l'inflammasome pyrine. Des mutations sur ces résidus sérines (S208, S242) peuvent entraîner une maladie différente de la FMF: la PAAND (Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis), une dermatose neutrophilique auto-inflammatoire [23].
Une inactivation des RhoA GTPase (par des toxines bactériennes par exemple) entraîne l'inactivation des kinases PKN1/PKN2 et la déphosphoration de la pyrine[24]. Chez les sujets sains, l'étape de déphosphoration seule n’entraîne pas l'activation de l'inflammasome pyrine. En revanche, chez les patients atteints de FMF, la déphosphorylation seule des sérines est suffisante pour déclencher l'activation de l'inflammasome pyrine[25]. Ceci suggère qu'il existe une régulation à deux niveaux et que le second mécanisme de régulation (indépendant de la (dé)phosphorylation) est déficient chez les patients FMF.
Ce mécanisme défaillant se situe vraisemblablement au niveau du domaine B30.2 (exon 10) où est située la plupart des mutations pathogènes associées à la FMF. C'est vraisemblablement l'interaction de ce domaine avec le cytosquelette (microtubules) qui est défaillante, comme le suggère l'efficacité de la colchicine[26]. Récemment deux types de signaux activateurs de l'inflammasome pyrine ont été mis en évidence, in vitro: des analogues d'acides biliaires (BAA 473/485 non présents physiologiquement dans l'organisme humain, mais possiblement métabolisés par le microbiote)[27] et des catabolites de la progestérone et de la testostérone (la prégnanolone et l'étiocholanolone) présents de manière physiologique dans l'organisme humain [28]. Ces derniers pourraient faire le lien entre le stress (car ce sont des neurostéroïdes), le cycle menstruel et les poussées de la maladie.
Les mutations à l'origine de la FMF abaissent le seuil d'activation de l'inflammasome pyrine[29]. Ces mutations pourraient avoir, à l'état hétérozygote, avoir conféré un avantage sélectif face aux épidémies historiques (notamment la Peste due à Yersinia pestis)[30], ce qui expliquerait sa plus forte prévalence dans le bassin méditerranéen.
Clinique
Présentation générale
Après une période de latence, qui dans la majorité des cas est brève et ne dépasse pas l’enfance, la maladie est caractérisée par la survenue d’accès aigus séparés par des périodes asymptomatiques de durée irrégulière, variant de quelques jours à plusieurs années.
La symptomatologie de la maladie périodique présente deux aspects : les manifestations paroxystiques, bruyantes mais d’évolution le plus souvent favorable spontanément, et l’amylose, complication chronique, dont la survenue éventuelle conditionne le pronostic.
Les symptômes de la maladie périodique surviennent une fois sur deux dans les 10 premières années de la vie et dans 90% des cas avant l’âge de 20 ans. Un début après l'âge de 30 ans est exceptionnel[31].
Dans la grande majorité des cas, l’affection commence par une crise aiguë abdominale, posant le problème d’une urgence de type chirurgical. Les accès apparaissent brusquement, atteignant leur acmé en quelques heures, et régressent habituellement en quelques jours.
Ils se répètent de manière totalement imprévisible, parfois déclenchés par certains facteurs : activité physique inhabituelle, exposition au froid, traumatisme, intervention chirurgicale, infection, alimentation riche en graisses, émotion ou période menstruelle[1]...
Accès fébrile
Il se traduit par une élévation brusque de la température qui atteint 38 à 39 °C (parfois 40 °C) en quelques heures. Ces crises fébriles durent en moyenne une demi-journée à trois jours. Cet accès peut être isolé, « pseudopalustre » ou accompagner une manifestation viscérale[4].
La fièvre s’atténue habituellement en 12 à 24 heures, mais peut persister jusqu’à 5 jours, voire plus longtemps, en particulier lorsqu’il existe une atteinte articulaire.
Ces crises s'accompagnent le plus souvent d'une inflammation des séreuses à l'origine des symptômes : péritonite et douleurs abominales (90 à 95% des cas), synovites et douleurs articulaires (20 à 70 % des cas), pleurésies et douleurs thoraciques (40%), plus rarement testiculaire (orchite) ou cardiaque (péricardite)[16].
Des éruptions cutanées peuvent également apparaître tandis que la présence d'une fièvre lors des crises est un bon indicateur de l'existence de cette maladie. Toutefois, la fièvre peut être absente dans 5 % des cas[32].
Accès péritonéal
Il est la manifestation la plus caractéristique et, avec la fièvre, le symptôme le plus fréquent de la maladie périodique (90 à 95% des cas)[1] - [16].
Il simule une urgence chirurgicale, avec parfois défense, voire véritable contracture pariétale, ou aspect radiographique évoquant une occlusion intestinale.
Lorsqu’un tel tableau est inaugural (lors de la première crise), le diagnostic est très difficile, l’intervention chirurgicale est presque inévitable. En l’absence d’intervention, la douleur commence à régresser après 6 à 12 heures et sa disparition, complète en 24 à 48 heures, s’accompagne souvent d’une diarrhée transitoire[1] - [16].
Le diagnostic différentiel avec une urgence chirurgicale est souvent très délicat, reposant sur une analyse sémiologique rigoureuse et sur une surveillance soigneuse. La prolongation de la crise pendant plus de 24 heures doit faire reconsidérer le diagnostic et renforcer la surveillance pour ne pas laisser passer l’heure d’une intervention chirurgicale.
Crises articulaires
Elles surviennent surtout chez l'enfant (dans 20 à 70% des cas), en particulier les grosses articulations, notamment le genou, la cheville, la hanche et l’épaule.
Il s’agit habituellement d’une monoarthrite, plus rarement d’une oligoarthrite ou d’une polyarthrite. Les accès articulaires peuvent se présenter sous deux formes.
Les accès aigus, les plus fréquents, réalisent un tableau d’arthrite, avec parfois épanchement fugace constitué d’un liquide d’aspect clair, trouble ou puriforme, contenant de 200 à 1 000 000 d’éléments par millimètre cube, polynucléaires neutrophiles non altérés essentiellement. La crise atteint son acmé en 2 à 3 jours, puis régresse en 1 semaine environ, le plus souvent sans aucune séquelle.
Les formes prolongées sont moins fréquentes et intéressent surtout le genou et la hanche. Le tableau est celui d’une monoarthrite chronique qui s’accompagne souvent d’une attitude en flexum et d’une déminéralisation osseuse, souvent importante. Les symptômes ne commencent à régresser qu’après un délai de plusieurs mois à 1 an et finissent par disparaître, le plus souvent sans séquelle.
Très rarement néanmoins, une arthropathie destructrice chronique peut se développer au genou et surtout à la hanche, compromettant alors le pronostic fonctionnel[16].
Accès thoraciques
Ils réalisent un tableau de pleurésie aiguë fébrile régressant totalement en 24 à 48 heures (40% des cas).
Signes cutanés
Ils se traduisent surtout par un érythème érysipéloïde (ou « pseudo-érysipèle ») siégeant aux membres inférieurs, ou par diverses autres lésions, avec parfois une vascularite[16]. .
Sont encore signalées la survenue d’une orchite aiguë unilatérale chez les garçons âgés de moins de 16 ans, et exceptionnellement d’une méningite périodique aseptique ou d’une péricardite[1] - [16].
Signes biologiques
Le diagnostic de la maladie périodique reste encore purement clinique car, malgré de nombreuses recherches, aucun marqueur biologique spécifique de l’affection n’a pu être mis en évidence[16].
Le caractère inflammatoire de la maladie doit être confirmé, dont l’augmentation de la vitesse de sédimentation globulaire, correspondant à une augmentation du fibrinogène et des alpha2-globulines, et le dosage de la CRP (Protéine C reactive). L'hyperleucocytose est moins constante[31].
L’enquête immunologique, et notamment la recherche d’autoanticorps, est habituellement négative[16].
L'absence d'inflammation au cours d'un accès douloureux élimine le diagnostic de maladie périodique[17].
Critères du diagnostic
Diagnostic clinique
En l’absence de marqueur biologique spécifique, le diagnostic de la maladie périodique repose sur une analyse sémiologique rigoureuse, confirmée éventuellement par un test thérapeutique par la colchicine.
Le diagnostic est évoqué devant un patient ayant des accès de fièvre à répétition accompagnés de symptômes caractéristiques, ayant débuté dans l'enfance, et appuyé par un syndrome inflammatoire biologique durant un accès aigu[31] - [17].
Différents ensembles de critères ont été proposés pour le diagnostic, la sévérité, ou la surveillance du traitement[33].
Confirmation génétique
Un test génétique peut permettre de détecter la présence des mutations responsables de la maladie. Il peut s'agir d'une ou deux mutations du gène MEFV, soit deux mutations identiques homozygotes (même mutation provenant des deux parents), soit deux mutations différentes hétérozygotes composites (chaque parent apporte une mutation différente). Mais l'absence de résultat n'élimine pas le diagnostic[31].
En effet, le résultat de l'analyse génétique doit être interprété en fonction du contexte de chaque patient[17], car des patients ayant des troubles caractéristiques peuvent être porteurs de mutations non encore identifiées[1] - [14] dans la base de données dédiée[34].
Dans les cas douteux, un test thérapeutique à la colchicine de 3 à 6 mois peut être utile pour trancher[31].
Évolution et complications
Le pronostic de la maladie périodique dépend essentiellement du risque d’amylose et a été complètement modifié par l’efficacité préventive de la colchicine.
Si l’amylose n’apparaît pas, le pronostic est relativement bon, et pour certains auteurs, la durée de survie serait identique à celle des sujets normaux.
Si un traitement n'est pas initié précocement, la principale complication à apparaître est la survenue d'une amylose rénale de type AA, conduisant à une insuffisance rénale et nécessitant le recours à la dialyse.
Amylose rénale
L’amylose rénale est de loin la plus précoce et la plus fréquente des complications
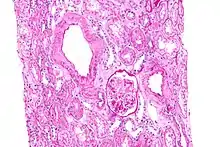
La substance amyloïde de la maladie périodique est formée de fibrilles identiques à la protéine de l'amylose AA, et elle intéresse de manière diffuse la paroi de toutes les artérioles, sauf celles du système nerveux central ; cette répartition est dominée par la localisation rénale..
La survenue d’une amylose au cours de la maladie périodique transforme une affection invalidante mais bénigne en une maladie mortelle quasi certaine. Son incidence est élevée chez les Juifs originaires d’Afrique du Nord et chez les Turcs et plus faible chez les Arméniens, les Arabes et les Juifs ashkénazes[1].
Son évolution passe par deux phases principales. Le début est asymptomatique, il s'agit d'une protéinurie modérée, de durée moyenne de 3 à 4 ans. Son apparition au cours de la maladie périodique constitue une très forte présomption d’amylose, et cette protéinurie doit être recherchée au moins une fois par an[31].
La phase néphrotique est caractérisée par l’apparition d’un syndrome néphrotique clinique et biologique. La confirmation histologique peut être apportée par la biopsie rénale, la biopsie rectale (positive dans 75 à 85 % des cas), voire par la biopsie médullaire.
Elle laisse place, après en moyenne 1 ou 2 ans d’évolution, à la phase urémique avec insuffisance rénale, qui progresse en règle rapidement, pour atteindre son stade terminal 12 à 18 mois plus tard.
Avant l’ère de l’hémodialyse, la durée de survie était en moyenne de 7 ans après l’apparition de la protéinurie et de 3 ans après celle de l’insuffisance rénale. Les patients atteints de maladie périodique et d’insuffisance au stade terminal sont de bons candidats à l’hémodialyse et à la greffe rénale.
Le risque essentiel est alors la poursuite du processus amyloïde dans les autres organes (cœur, intestin et surrénales notamment) et sa récidive éventuelle sur le greffon, complications dont la survenue pourrait être évitée ou freinée par la poursuite du traitement par la colchicine.
Autres complications
Une péritonite chronique peut se cloisonner donnant une ascite ou des kystes. Des arhroses ou arthrites parfois destructives peuvent apparaitre au niveau de la hanche ou de la colonne vertébrale.
Le caractère chronique de la maladie et imprévisible des accès inflammatoires retentit sur la qualité de vie, avec des conséquences sociales (absentéisme scolaire ou professionnel).
Chez la femme enceinte, la plupart des auteurs notaient, avant la découverte de l'efficacité de la colchicine, une diminution des crises durant la grossesse[14]. Cependant d'autres auteurs ont mis en évidence un plus grand risque de complications de la grossesse chez les femmes atteintes de maladie périodique (avortements spontanés en début de grossesse, naissances prématurées...)[16] - [31].
Traitement
Traitement de fond

Depuis 1972[35], l’immense majorité des cas de maladie périodique est traitée de manière continue par la colchicine. Ce traitement est débuté dès que le diagnostic est confirmé ou fortement suspecté, et poursuivi à vie[31].
La dose de départ varie selon l'âge et l'état du sujet. L'efficacité est évaluée sur le nombre et la durée des épisodes aigus.et une éventuelle inflammation résiduelle. La posologie habituelle est de l’ordre de 1 à 2 mg/j en 1 ou 2 prises, quels que soient le poids et l’âge[16].
Cette méthode thérapeutique permet, dans la grande majorité des cas, de faire disparaître totalement, ou tout au moins d’espacer, les accès. Il est largement utilisé même si le mécanisme de cette thérapie reste incompris.
Résultats
La colchicine n'a pas d'activité sur une crise aiguë en cours, même en augmentant les doses. On adjoint un traitement symptomatique anti-inflammatoire et antalgique[31].
Le traitement continu par la colchicine prévient, ou tout au moins retarde, l’apparition de l’amylose dans l’immense majorité des cas, même lorsqu’il reste sans effet sur la répétition des accès paroxystiques[17].
L’action curative de la colchicine vis-à-vis d’une amylose déjà déclarée est moindre, mais peut se traduire par la stabilisation, la régression et même parfois la disparition de la protéinurie, à condition que celle-ci ne soit pas trop évoluée et que des posologies supérieures à 1,5 mg/j soient utilisées[17].
Effets secondaires
L'innocuité de la colchicine a été montrée chez les enfants (courbe de croissance normale) et chez la femme enceinte (à doses habituelles). L'allaitement est possible. Le principal effet secondaire est la diarrhée, qui disparait en général spontanément en deux à trois semaines[31].
Les risques de surdosage, avec effets toxiques, apparaissent surtout lors de la survenue d'une insuffisance rénale, ou en interaction médicamenteuse (comme les antibiotiques de type macrolides)[17].
Traitement des formes résistantes
Dans 10 % des cas, même lorsque le traitement est scrupuleusement suivi, il existe une résistance à la colchicine (inefficacité à la dose maximale). Le traitement est alors mal codifié, reposant sur les biothérapies (inhibiteur de l'interleukine 1 comme le canakinumab ou l'anakinra, anti-TNF...)[16].
Le canakinumab a été commercialisé sous le nom « Ilaris » ou ACZ885, par Novartis Pharma. Cette molécule agit en bloquant une protéine dite interleukine-1 bêta, laquelle joue un rôle dans les processus inflammatoires. Cette prescription se fait en collaboration avec un centre référent pour la maladie[31].
Références
- « Familial Mediterranean Fever »,
- « Maladie périodique de Reimann », sur journals.lww.com (consulté le )
- Mamou H. et Cattan R., « La maladie périodique », Semaine des Hôpitaux de Paris, no 28, , p. 1062-1070.
- Raoul Ghozlan, « Maladie périodique », Tempo médical, no 467, , p. 29
- Garnier Delamare, Dictionnaire des termes de médecine, Maloine, (ISBN 2-224-02381-2), p. 305.
- Henry Mamou, La maladie périodique, Expansion scientifique française, , 169 p.
- A. Manuila, Dictionnaire français de médecine et de biologie, vol. II, Masson, , p. 62 et 726-727.
- Garnier Delamare, Dictionnaire illustré des termes de médecine, Paris, Maloine, , 1094 p. (ISBN 978-2-224-03434-4), p. 721.
- S. E. Goldfinger, « Colchicine for familial Mediterranean fever », The New England Journal of Medicine, vol. 287, no 25, , p. 1302 (ISSN 0028-4793, PMID 4636899, DOI 10.1056/NEJM197212212872514, lire en ligne, consulté le )
- « Des fièvres récurrentes héréditaires aux syndromes auto-inflammatoires : les apports de la génétique – Académie nationale de médecine | Une institution dans son temps » (consulté le )
- André Julien Fabre, « Le colchique : deux millénaires d'actualité », Histoire des Sciences médicales, vol. 39, no 2, , p. 149. (lire en ligne)
- (en) L Gruberg, I Aksentijevich, E Pras, D L Kastner et M Pras, « Mapping of the familial Mediterranean fever gene to chromosome 16 », American journal of reproductive immunology (New York, N.Y.: 1989), vol. 28, nos 3-4, , p. 241-242 (ISSN 1046-7408, PMID 1285890)
- V. Lemaire, « Le gène de la maladie périodique a été cloné », Le Concours Médical, vol. 120, no 20, , p. 1397
- Société française de génétique humaine, « Fièvre méditerranéenne familiale », La Revue du Praticien, vol. 53, , p. 820-824.
- Gilles Grateau, « Fièvres récurrentes héréditaires », Le Concours Médical, vol. 126, no 21, , p. 1207-1212
- İsmail Sarı, Merih Birlik et Timuçin Kasifoğlu, « Familial Mediterranean fever: An updated review », European Journal of Rheumatology, vol. 1, no 1, , p. 21–33 (ISSN 2147-9720, PMID 27708867, PMCID PMC5042258, DOI 10.5152/eurjrheum.2014.006, lire en ligne, consulté le )
- Gilles Grateau, « Fièvre méditerranéenne familiale », La Revue du Praticien, vol. 61, , p. 1349-1350.
- Gilles Grateau, « Les syndromes auto-inflammatoires », La Revue du Praticien, vol. 55, , p. 353-359
- (en) French FMF consortium - Group 1:Alain Bernot, Christian Clepet, Corinne Dasilva, Catherine Devaud, Jean-Louis Petit, Christophe Caloustian, Corinne Cruaud, Delphine Samson, Françoise Pulcini, Jean Weissenbach & Roland Heilig Group 2: Cécile Notanicola, Cécile Domingo, Michael Rozenbaum, Eldad Benchetrit, Rezzan Topaloglu, Marie Dewalle, Christiane Dross, Philippe Hadjari, Madeleine Dupont, Jacques Demaille & Isabelle Touitou Group 3: Nizar Smaoui, Brigitte Nedelec, Jean-Philippe Méry, Habiba Chaabouni, Marc Delpech & Gilles Grateau, « A candidate gene for familial Mediterranean fever », Nature Genetics, vol. 17, no 1, , p. 25-31 (PMID 9288094, DOI 10.1038/ng0997-25, lire en ligne, consulté le )
- (en) The International FMF Consortium, « Ancient Missense Mutations in a New Member of the RoRet Gene Family Are Likely to Cause Familial Mediterranean Fever », Cell, vol. 90, no 4, , p. 797-807 (ISSN 0092-8674, PMID 9288758, DOI 10.1016/S0092-8674(00)80539-5, lire en ligne, consulté le )
- Jae Jin Chae, Young-Hun Cho, Geun-Shik Lee et Jun Cheng, « Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice », Immunity, vol. 34, no 5, , p. 755–768 (ISSN 1097-4180, PMID 21600797, PMCID 3129608, DOI 10.1016/j.immuni.2011.02.020, lire en ligne, consulté le )
- Yvan Jamilloux, Flora Magnotti, Alexandre Belot et Thomas Henry, « The pyrin inflammasome: from sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes », Pathogens and Disease, vol. 76, no 3, 04 01, 2018 (ISSN 2049-632X, PMID 29718184, DOI 10.1093/femspd/fty020, lire en ligne, consulté le )
- Fiona Moghaddas, Rafael Llamas, Dominic De Nardo et Helios Martinez-Banaclocha, « A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to Familial Mediterranean Fever », Annals of the Rheumatic Diseases, vol. 76, no 12, , p. 2085–2094 (ISSN 1468-2060, PMID 28835462, PMCID 5687562, DOI 10.1136/annrheumdis-2017-211473, lire en ligne, consulté le )
- Yong Hwan Park, Geryl Wood, Daniel L. Kastner et Jae Jin Chae, « Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS », Nature Immunology, vol. 17, no 8, , p. 914–921 (ISSN 1529-2916, PMID 27270401, PMCID 4955684, DOI 10.1038/ni.3457, lire en ligne, consulté le )
- Flora Magnotti, Lucie Lefeuvre, Sarah Benezech et Tiphaine Malsot, « Pyrin dephosphorylation is sufficient to trigger inflammasome activation in familial Mediterranean fever patients », EMBO Molecular Medicine, vol. 11, no 11, 11 07, 2019, e10547 (ISSN 1757-4684, PMID 31589380, PMCID 6835204, DOI 10.15252/emmm.201910547, lire en ligne, consulté le )
- Hanne Van Gorp, Pedro H. V. Saavedra, Nathalia M. de Vasconcelos et Nina Van Opdenbosch, « Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation », Proceedings of the National Academy of Sciences of the United States of America, vol. 113, no 50, 12 13, 2016, p. 14384–14389 (ISSN 1091-6490, PMID 27911804, PMCID 5167202, DOI 10.1073/pnas.1613156113, lire en ligne, consulté le )
- Irina Alimov, Suchithra Menon, Nadire Cochran et Rob Maher, « Bile acid analogues are activators of pyrin inflammasome », The Journal of Biological Chemistry, vol. 294, no 10, , p. 3359–3366 (ISSN 1083-351X, PMID 30647128, PMCID 6416436, DOI 10.1074/jbc.RA118.005103, lire en ligne, consulté le )
- (en) Flora Magnotti, Daria Chirita, Sarah Dalmon et Amandine Martin, « Steroid hormone catabolites activate the pyrin inflammasome through a non-canonical mechanism », BioRxiv (prépublication), Immunology, (DOI 10.1101/2021.10.29.466454, lire en ligne, consulté le )
- Yvan Jamilloux, Lucie Lefeuvre, Flora Magnotti et Amandine Martin, « Familial Mediterranean fever mutations are hypermorphic mutations that specifically decrease the activation threshold of the Pyrin inflammasome », Rheumatology (Oxford, England), vol. 57, no 1, 01 01, 2018, p. 100–111 (ISSN 1462-0332, PMID 29040788, DOI 10.1093/rheumatology/kex373, lire en ligne, consulté le )
- Lawton K. Chung, Yong Hwan Park, Yueting Zheng et Igor E. Brodsky, « The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome », Cell Host & Microbe, vol. 20, no 3, , p. 296–306 (ISSN 1934-6069, PMID 27569559, PMCID 5025386, DOI 10.1016/j.chom.2016.07.018, lire en ligne, consulté le )
- Katia Stankovic Stojanovic, « Fièvre méditerranéenne familiale », La Revue du Praticien - médecine générale, , p. 768-769.
- Z. Amoura, B. Wechsler, « Maladie périodique », sur www.therapeutique-dermatologique.org, (consulté le )
- (en) Mordechai Shohat, Familial Mediterranean Fever, University of Washington, Seattle, (PMID 20301405, lire en ligne)
- Base de données infevers
- (en) SE Goldfinger, « Colchicine for Familial Mediterranean Fever », New England Journal of Medicine, vol. 287, no 25, , p. 1302-1302 (ISSN 0028-4793, PMID 4636899, DOI 10.1056/NEJM197212212872514, lire en ligne, consulté le )