Ensemble canonique
En physique statistique, l’ensemble (ou situation) canonique est un ensemble statistique introduit par le physicien américain Josiah Willard Gibbs[1]. Il correspond au cas d'un système physique de volume donné et contenant un nombre fixe de particules, en interaction avec un autre système, appelé réservoir ou thermostat, beaucoup plus grand que le système considéré et avec lequel il peut échanger de l'énergie mais pas de matière. Le thermostat se comporte comme un réservoir supposé infini d'énergie, la réunion des deux systèmes étant considérée comme isolée. Le couplage entre le système étudié et le réservoir est considéré comme faible, c'est-à-dire que l'état du réservoir n'est pas modifié quels que soient les échanges d'énergie entre lui et le système[2].


Un exemple d'une telle situation peut être donné par une bouteille d'eau fermée et plongée dans une piscine : cette dernière constitue le réservoir. Il est clair que même si la bouteille est initialement à une température beaucoup plus basse, ou plus élevée, que celle de la piscine, elle n'influencera pas de façon mesurable la température de la piscine[n 1]. La notion de réservoir est relative, ainsi une tasse de thé chaud pourra être approximativement considérée comme un réservoir pour une tranche de citron plongée dedans[n 2] mais certainement pas pour toute la pièce dans laquelle elle se trouve, qui pourra à l'inverse être considérée comme le réservoir vis-à-vis du système constitué par la tasse de thé (et la tranche de citron)[n 3].
La condition d'équilibre thermique entre le système étudié et le réservoir est réalisée lorsqu'ils sont à la même température. Plus précisément, le réservoir, beaucoup plus gros que le système considéré, impose sa température à ce dernier : c'est pourquoi il est souvent appelé thermostat.
L'ensemble canonique est l'ensemble des « copies virtuelles » du même système dans le même état d'équilibre avec le thermostat, donc à la même température. Contrairement au cas de l'ensemble microcanonique, l'énergie du système étudié est alors amenée à fluctuer d’une « copie » du système à une autre de l'ensemble. Toutefois, et contrairement à la situation microcanonique, les différents micro-états d'énergie du système étudié ne possèdent pas tous la même probabilité, du fait de l'interaction avec le réservoir. Il est possible de déterminer la forme générale de la distribution de probabilité des micro-états d'énergie accessibles du système, appelée distribution canonique.

Distribution canonique
Dans la situation canonique, le volume et le nombre de particules du système correspondent à des paramètres extérieurs[n 4] (en l'espèce, extensifs), dont les valeurs sont fixées. Il convient de souligner que la fixité du volume du système implique que celui-ci n'échange pas de travail avec le réservoir : les échanges d'énergie avec ce dernier sont purement thermiques[3] - [n 5]. Par ailleurs elle peut être difficile à réaliser en pratique, du fait des phénomènes de dilatation-contraction des parois du récipient contenant le système: de fait cette condition de volume fixe est rarement réalisée en chimie par exemple, où l'on préfère considérer que c'est la pression qui est fixe[n 6].
En revanche, et contrairement au cas microcanonique, l'énergie du système n'est plus fixée, en raison des échanges avec le réservoir, et est donc une variable interne, au même titre que peut l'être par exemple la densité de la matière constituant le système. Par suite, la distribution des différents micro-états du système n'a aucune raison d'être celle de l'ensemble microcanonique, où ceux-ci sont équiprobables, mais doit être déterminée en prenant en compte ces échanges système - réservoir.

La détermination de la distribution des niveaux d'énergie au sein d'un ensemble canonique se fait en considérant l'ensemble microcanonique qu'il constitue avec le réservoir d'énergie. L'énergie totale du système étudié et du réservoir est notée , celle du système dans un micro-état donné ( étant un indice générique permettant de distinguer les divers micro-états), et celle correspondante du réservoir , avec dans tous les cas . L'ensemble {système étudié + réservoir} étant considéré comme isolé, l'énergie est constante à près, et pour que le thermostat constitue effectivement un réservoir infini d'énergie pour le système considéré.






Condition d'équilibre thermique
À l'équilibre thermodynamique l'entropie de l'ensemble {système + réservoir} doit être maximale. Comme l'entropie est une grandeur extensive, l'entropie de l'ensemble {système + réservoir} est la somme des entropies du système, notée , et du réservoir, notée :



- .

Le système n'interagissant avec le réservoir qu'en s'échangeant une énergie avec ce dernier, il est possible d'écrire la variation correspondante de l'entropie totale[n 7] :
- .

À l'équilibre thermique l'entropie de l'ensemble {système + réservoir} est maximale donc , par suite il vient :

- .

Par ailleurs, comme en pratique , où est la température microcanonique du réservoir, qui ne dépend donc pas de l'état du système étudié. Il est alors possible de poser , étant la température canonique du système étudié, et la relation d'équilibre thermique entre ce dernier et le réservoir se réduit à . Le réservoir "impose" donc à l'équilibre sa température, supposée constante, au système étudié, d'où le terme souvent utilisé de thermostat pour le désigner.





Tout ceci suggère d'introduire le facteur :

Ceci constitue pour ainsi dire une définition du concept de température. Lorsque ces deux facteurs sont égaux, les variations d'entropies seront strictement opposées pour les deux systèmes puisqu'ils échangent une quantité d'énergie toujours strictement opposée. À température égale, on est donc assuré du caractère extrémal (mais en fait bel et bien maximal) de l'entropie, ce qui assure l'équilibre thermodynamique.
Distribution des micro-états du système
Soient le nombre de micro-états du système total d'énergie totale , constante (à près), et celui du réservoir lorsque le système étudié est dans un micro-état d'énergie . Comme lorsque le système se trouve dans ce micro-état le réservoir peut se trouver dans n'importe lequel des micro-états correspondants, sans qu'aucun ne soit favorisé, la probabilité de trouver le système étudié dans un micro-état donné d'énergie au sein de l'ensemble canonique est donc donnée par :



- ,

c'est-à-dire le nombre de cas favorables sur le nombre total de cas possibles[n 8]. Le dénominateur n'étant qu'une constante indépendante du micro-état considéré, il est possible d'écrire en passant au logarithme et faisant apparaître l'entropie microcanonique du réservoir :

- ,

soit encore en tenant compte de la condition d'équilibre thermique entre le système et le réservoir :

- .

Il vient dès lors l'expression de la probabilité de trouver le système dans le micro-état :
- ,

où est une constante de normalisation donnée par la condition , la sommation portant sur tous les micro-états possibles du système[n 9].


Cette expression peut dès lors se réécrire sous la forme suivante[n 10] :

où est la fonction de partition canonique définie par[n 10] :


Cas continu
Dans le cas où le spectre des micro-états du système peut être considéré comme continu ou quasi-continu[n 11], il est fréquent de recourir à une description continue. On introduit alors la notion de densité d'états telle que le nombre de micro-états du système d'énergies comprises entre et soit donné par :



- .

Il faut alors considérer la densité de probabilité pour que les micro-états du système aient des énergies comprises entre et , qui est alors donnée selon la formule précédente modifiée sous la forme :

- ,

la fonction de partition étant alors donnée par :
- ,

l'intégrale portant sur l'ensemble des valeurs possibles de l'énergie[n 12].
Discussion physique
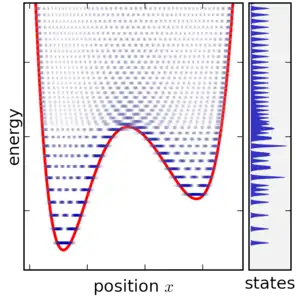
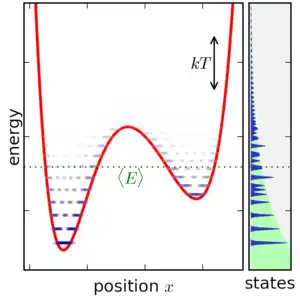

Contrairement au cas du formalisme microcanonique, les divers micro-états du système étudié ne sont pas équiprobables. Si on considère deux valeurs distinctes de l'énergie, notées et , avec , le rapport de leurs probabilités au sein de l'ensemble canonique, qui correspond au rapport de leurs "populations" respectives, est donné par :



- , avec .


Clairement plus l'écart énergétique entre les niveaux est important pour une température donnée, plus le ratio de population sera faible, l'état de plus basse énergie étant le plus probable[n 13].

Le ratio de probabilité des différents états est « modulé » par la quantité , qui joue le rôle d'une énergie caractéristique[3], traduisant en quelque sorte l'influence de "l'agitation thermique" (lié au réservoir) sur l'occupation des différents micro-états du système en fonction de leurs énergies respectives.

Il est utile de retenir l'ordre de grandeur suivant[3] :

Par suite, l'écart énergétique entre les différents niveaux d'énergie d'un atome étant de l'ordre de l'électron-volt, il est extrêmement improbable de trouver les atomes d'un gaz monoatomique comme l'hélium ou le néon dans un état excité à la température ordinaire = 300 K, puisque pour le ratio précédent est de l'ordre de 4 × 10−18. On dit alors que les « degrés de liberté électroniques » sont gelés à la température ordinaire[n 14]. En revanche, pour des températures élevées, de l'ordre de plusieurs milliers de degrés, les états excités pourront jouer un rôle important.

À l'inverse, dans le cas de gaz constitués de molécules, il existe des degrés de libertés associés aux vibrations et aux rotations moléculaires. Typiquement, les écart d'énergies associés à ces termes sont de l'ordre de 0,1 à 0,01 eV et 10−3 – 10−5 eV, respectivement[4]. Ainsi les degrés de liberté de rotation ne seront "gelés" qu'à très basse température, et joueront certainement un rôle à température ordinaire, et il en sera de même pour ceux de vibration dès lors que la température est suffisamment élevée[n 15].
Il faudra donc tenir compte de ces degrés de liberté supplémentaires, ce qui explique pourquoi l'expression de l'énergie interne d'un gaz parfait varie selon que l'on considère un gaz monoatomique, pour lesquels il n'y a pas de degrés de liberté de rotation ou de vibration, et est alors donnée par (par mole), ou un gaz diatomique, où il faut alors tenir compte des deux degrés de liberté supplémentaires liés à la rotation, ce qui donne . À haute température, cette valeur tendra vers du fait du "dégel" des degrés de liberté de vibration[5].



Autre approche pour obtenir la distribution canonique
Il est possible de procéder différemment pour obtenir la distribution précédente, en utilisant une approche « généralisée »[6] - [7] - [8] du postulat fondamental de la physique statistique. Si cette approche est fortement calculatoire, par rapport à celle plus physique employée plus haut, elle est très féconde, et présente l'avantage de pouvoir être généralisée à pratiquement n'importe quelle situation.
Forme « généralisée » du postulat fondamental
En premier lieu, il convient de rappeler le postulat fondamental de la physique statistique :
- Étant donné un système isolé en équilibre, il se trouve avec des probabilités égales dans chacun de ses micro-états accessibles[9].
Ce postulat privilégie évidemment les systèmes isolés, et son application à la situation canonique implique de raisonner sur le système global, isolé, et effectuant des hypothèses de faible couplage sur le système étudié, pour y avoir recours.
Il est possible de généraliser ce postulat de la façon suivante :
- À l'équilibre macroscopique, la distribution statistique des états microscopiques est celle qui maximise l'entropie statistique du système , compte tenu des contraintes extérieures qui sont imposées à ce dernier[6] - [8].

Dans tous les cas la distribution de probabilité devra respecter la contrainte liée à la condition de normalisation , qui peut aussi être mise sous la forme :

- .

Par ailleurs, dans le cas de l'ensemble canonique, la seconde contrainte qui porte sur la distribution de probabilité est que la valeur moyenne de l'énergie du système étudié est fixée à l'équilibre de par son contact avec le réservoir (supposé infini) d'énergie[8], c'est-à-dire qu'il faut que soit telle que :


- [6].

Application à l'ensemble canonique
Le problème de la détermination de la distribution de probabilité dans le cas de l'ensemble canonique est donc celui d'une maximisation sous contrainte de la quantité , les deux contraintes étant :

- , (condition de normalisation) ;
- , (contact avec le réservoir d'énergie).

Ce problème peut se résoudre en utilisant la méthode des multiplicateurs de Lagrange: on cherche alors à maximiser la quantité[6]:
- ,

et étant deux constantes arbitraires qui seront déterminées ultérieurement en remplaçant l'expression obtenue de dans celles des deux contraintes imposées à la distribution.



On minimise alors en considérant chacun des comme une variable indépendante, c'est-à-dire en cherchant tel que , ce qui donne les conditions :


- ,

ce qui implique :
- , avec constante de normalisation.

On obtient bien une distribution de la même forme que précédemment, la constante étant bien l'inverse de la température comme il est possible de le vérifier en substituant cette expression dans celle de l'entropie statistique . Il vient en effet après réarrangement :

- ,

soit encore compte tenu du fait que et de :


- .

Il suffit de remarquer que où est la température du réservoir imposée au système pour vérifier que .


L'utilisation de la contrainte permet alors de montrer que .

Expression des fonctions thermodynamiques
La fonction de partition canonique ne dépend pas de l'énergie des micro-états individuels, en revanche elle dépend en général des paramètres extérieurs du système, notamment sa température et son volume . On vérifie que permet de déterminer simplement les principales caractéristiques du système, dont son énergie moyenne et son entropie statistique , lesquelles s'identifient à la limite thermodynamique à l'énergie interne et à l'entropie du système étudié[3].


Énergie moyenne du système - Énergie interne
Par définition, l'énergie moyenne du système est donnée par la relation[3] - [2] :
- ,

or il est facile de vérifier l'identité :
- ,

soit encore en repassant à la température :
- .

À la limite thermodynamique, s'identifie à l'énergie interne du système.
Entropie du système
Il est possible d'établir l'expression de l'entropie du système étudié dans la situation canonique en utilisant la définition de l'entropie statistique sur un ensemble :
- ,
en substituant dans cette expression celle de correspondant à la distribution canonique, il vient :
- ,

où il a été tenu compte du fait que d'après la définition de la fonction de partition .

Compte tenu de l'expression précédente de il est possible de réécrire l'expression de l'entropie statistique du système sous la forme :
- .

À la limite thermodynamique, s'identifie à l'entropie thermodynamique du système.
Énergie libre
Les expressions précédentes de l'énergie interne et de l'entropie thermodynamique peuvent être réécrites sous une forme plus exploitable en introduisant l'énergie libre du système, définie à partir de la fonction de partition par :

- , soit encore .


On peut alors vérifier que l'énergie interne et l'entropie du système sont données en fonction de l'énergie libre par :



- ,

et
- ,

ce qui permet de retrouver l'identité classique: .

Notes et références
Notes
- En toute rigueur, il faudrait considérer que le volume de la bouteille est fixe, et donc négliger les phénomènes de dilatation ou de contraction de celle-ci.
- Cette image est due à Reif, op. cit., chapitre 3. Il convient de tenir compte du temps dans cette situation : initialement, la tasse de thé, très chaude, va imposer « rapidement », c'est-à-dire avant qu'elle n'ait le temps de refroidir, sa température à la tranche de citron. Mais ensuite, elle refroidira pour arriver à l'équilibre avec la pièce dans laquelle elle se trouve.
- À condition, en toute rigueur, de négliger les pertes par évaporation de la tasse de thé, pour ne pas avoir d'échange de matière.
- Comme c'est le cas de l'énergie dans la situation microcanonique et ne sont connus qu'aux incertitudes et près.
- On peut bien sûr "relâcher" ce paramètre extérieur, en faisant varier le volume, puis en laissant le système évoluer dans un nouvel état d'équilibre dans la situation canonique, avec cette nouvelle valeur de volume. Il y aura bien sûr échange de travail lors de ce "relâchement" du volume. Ce qu'il faut retenir est que dans la situation canonique "habituelle", le volume n'est pas contrairement à l'énergie une grandeur qui peut varier librement par échange avec un "réservoir de volume", de façon à parvenir à l'équilibre.
- Il est possible de considérer l'ensemble dit pour lequel le système est en contact avec un réservoir d'énergie et de volume, dans ce cas devient une variable interne et c'est la pression qui est imposée au système par le réservoir.
- En toute rigueur, on a et il conviendrait d'écrire les dérivées partielles sous la forme , etc. Toutefois pour alléger l'écriture les autres variables seront omise dans les dérivées partielles.
- Cette relation suppose en fait l'indépendance statistique entre les états des deux sous-systèmes constitués respectivement par le système considéré et le réservoir, qui est elle-même valable si ceux-ci sont faiblement couplés. Cf. Lev Landau et Evgueni Lifchits, Physique théorique, t. 5 : Physique statistique [détail des éditions], § 2 ss.
- Il convient de distinguer cette sommation, par état du système, de celle qui peut être effectuée par niveau d'énergie. Dans le premier cas la somme comporte un terme par état du système, et donc plusieurs états dégénérés, c'est-à-dire des états distincts mais de même énergie , apparaissent plusieurs fois dans la somme. Dans le second cas au contraire, il y a un terme dans la somme par valeur distincte de , il ce cas il faudrait tenir compte du degré de dégénérescence de chaque état dans les sommations. Cf. Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Éléments de physique statistique, [détail de l’édition], chapitre III, §I c.1.a.
- Dans le cas d'une somme par niveau d'énergie (cf. note précédente à ce sujet), il faudrait introduire le degré de dégénérescence de chaque état dans ces expressions, qui deviennent alors et , la notation rappelant que l'on doit sommer un terme par valeur de l'énergie et non par état, même dégénéré, du système. Cf. Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Éléments de physique statistique, [détail de l’édition].
- En pratique, il faut que les écarts entre deux niveaux d'énergie voisins soient très faibles devant l'énergie caractéristique liée à l'agitation thermique.
- Ceci peut paraître quelque peu contradictoire avec les hypothèses relatives au thermostat, car on autorise potentiellement des variations très grandes, voire infinies, de l'énergie, sans compter que certains de ces états peuvent être incompatibles avec les paramètres extérieurs imposés au système. En pratique cependant les états d'énergie ayant des valeurs extrêmes par rapport à la valeur moyenne sont très peu probables et ne contribuent pratiquement pas à l'intégrale.
- Attention, il est erronée en général de dire que la formule donnant implique que les énergies les plus basses soient les plus probables, car cette formule ne tient pas compte de éventuelles dégénérescences des différents états d'énergie, donc du nombre d'états associés à une valeur donnée de l'énergie, cf. note précédente.
- Toutefois, les sous-niveaux associés aux structures fine et hyperfine de l'état fondamental, lorsqu'ils sont présents, possèdent des écarts bien plus faibles, de l'ordre de 0,1–0,01 eV et 10−6 eV, respectivement, ce qui correspond dans le cas de la structure fine à des températures de l'ordre de quelques centaines à quelques milliers de K. Il peut alors s'avérer nécessaire de tenir compte de ces niveaux dans le calcul de la fonction de partition . Cependant, il est fréquent que la température soit suffisamment élevée pour que les différents sous-niveaux de structure fine, lorsqu'ils existent à l'état fondamental, soient tous excités et il est possible de raisonner comme s'ils étaient confondus en un seul niveau dégénéré, en utilisant le facteur de dégénérescence associé. Il en est de même pour l'éventuelle structure hyperfine, sauf à très basse température. Voir Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Éléments de physique statistique, [détail de l’édition], complément III.B, § II.1.
- Plus précisément, il est possible de définir des « températures caractéristiques » associées aux écarts entre les différents sous-niveaux. Ainsi par exemple l'écart entre deux niveaux de vibration d'une molécule diatomique, ou de deux niveaux d'un mode de vibration d'une molécule polyatomique, sera donné (dans l'approximation harmonique) par la quantité , liée à la "raideur" de l'oscillateur. Il est alors possible d'introduire la température caractéristique en posant . Cette température est typiquement de l'ordre de quelques milliers de K pour de nombreux gaz diatomique, mais elle peut être aussi basse que quelques centaines de K pour des gaz comme le dichlore ou le dibrome. Voir Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Éléments de physique statistique, [détail de l’édition], complément III.B § II.2 .













Références
- Josiah Willard Gibbs, Elementary Principles in Statistical Mechanics, New Tork, Charles Scribner's Sons, (lire en ligne).
- (en) Reif, Fundamentals of statistical and thermal physics (relié), New York, Mc Graw Hill, coll. « McGraw Hill series in fundamental of physics », (1re éd. 1965), 651 p., relié (ISBN 0-07-051800-9, LCCN 63022730), chap. 6.
- Voir Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Éléments de physique statistique, [détail de l’édition], chapitre III.
- Voir par exemple (en) Atkins, Elements of physical chemistry (relié), New York, Freeman and company, (1re éd. 1978), relié (ISBN 0-7167-2402-2, LCCN 93048911), chap. 16.
- Voir notamment Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Éléments de physique statistique, [détail de l’édition], complément III.B.
- (en) E. T. Jaynes, « Information theory and statistical mechanics », Physical Review, vol. 106, no 4, , p. 620-630 (DOI 10.1103/PhysRev.106.620, lire en ligne).
- (en) E. T. Jaynes, « Information theory and statistical mechanics II », Physical Review, vol. 108, no 2, , p. 171-190 (DOI 10.1103/PhysRev.108.171, lire en ligne).
- Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Éléments de physique statistique, [détail de l’édition], complément V.D, §I et § II.
- Cf. notamment Diu et al., op. cit., chapitre II, I, § B.1.b.
Bibliographie
![]() : document utilisé comme source pour la rédaction de cet article.
: document utilisé comme source pour la rédaction de cet article.
- (en) Josiah Willard Gibbs, Elementary principles in statistical mechanics, [détail de l’édition] (lire en ligne).
- Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Éléments de physique statistique, [détail de l’édition].

- Herbert G. Callen, Thermodynamics & an introduction to Thermostatistics, John Wiley & Sons, 2e édition, 1985, 494 p. (ISBN 0-471-86256-8).
- Lev Landau et Evgueni Lifchits, Physique théorique, t. 5 : Physique statistique [détail des éditions]
- (en) Gregory H. Wannier, Statistical Physics, Dover Publications Inc., 1987 (ISBN 0-486-65401-X).