Cristallisation d'un polymère
La cristallisation d'un polymère est une cristallisation observée dans certains polymères thermoplastiques. Lorsque la masse fondue d'un polymère se solidifie, il se produit un ordonnancement partiel des chaînes moléculaires dans ce polymère. À partir des noyaux de cristallisation, les chaînes moléculaires s’empilent sous forme de plis et forment ce que l'on appelle des lamelles. Ce sont les blocs de construction à partir desquelles d'autres unités structurelles telles que les sphérulites sont formées. En plus de la solidification, la cristallisation peut également avoir lieu à partir d'une solution ou par étirement.
Des exemples de polymères semi-cristallins sont le polyéthylène linéaire (PE), le polytétrafluoroéthylène (PTFE) et le polypropylène isotactique (PPi).
La formation de cristallites dépend des conditions de refroidissement, des additifs et des charges mélangés au polymère, ainsi que des conditions d'écoulement pendant la solidification.
La cristallisation influence les propriétés optiques, mécaniques, thermiques et chimiques du polymère. Le taux de cristallinité peut être mesuré par diverses méthodes analytiques. Cependant, les propriétés sont déterminées non seulement par le taux de cristallinité, mais aussi par la taille des unités structurelles ou l'orientation moléculaire.
Mécanismes de cristallisation
De nombreux phénomènes entourant la cristallisation des matériaux polymères ne sont pas encore entièrement compris ou même prouvés. Divers modèles ont été soutenus par des résultats expérimentaux et ont prévalu[1].
Cristallisation pendant la solidification de la masse fondue

Tous les polymères sont constitués de très longues chaînes macromoléculaires. Les polymères thermoplastiques sont caractérisés par le fait qu'ils se ramollissent ou fondent lorsque la température augmente. Dans la masse fondue, les chaînes macromoléculaires sont disposées irrégulièrement sous la forme de pelotes (figure 1). Ces chaînes s'interpénètrent de diverses manières (enchevêtrement). Dans le cas de nombreux polymères thermoplastiques, ce désordre reste sous forme d'une structure amorphe à l'état solide[2].
Lorsque la masse fondue d'un polymère semi-cristallin (un sous-groupe des thermoplastiques) est refroidie, les chaînes se déplacent de moins en moins et commencent à s'ordonner (cristallisation). Il s'agit d'une formation de cristallites avec une taille typique de 15–100 nm.
Dans les polymères semi-cristallins, des parties des chaînes macromoléculaires sont ordonnées les unes par rapport aux autres en parallèle. L'énergie serait plus favorable si les molécules étaient disposées parallèlement sur toute la longueur de la chaîne macromoléculaire. Cependant, étant donné que les chaînes macromoléculaires dans la masse fondue sont présentes sous la forme d'enchevêtrements confus, cet ordre ne peut en réalité être atteint ou ne peut être atteint qu'à très haute pression. Par conséquent, les cristallites se forment à partir de chaînes macromoléculaires repliées (figure 1), qui forment les structures de base d'unités structurelles plus grandes, telles que par exemple, des structures lamellaires. L'ordre ne doit pas être considéré comme complet. Il peut être formé de feuilles pliées avec des boucles de différentes dimensions. Les extrémités de la chaîne peuvent également être en désordre. De plus, il est courant qu'une chaîne macromoléculaire fait partie de deux cristallites. Chaque cristallite est donc constituée de sous-régions ordonnées (cristallines) et désordonnées (amorphes). C'est également la raison pour laquelle, même dans le cas où le polymère n'a pas de régions amorphes visibles macroscopiquement, un matériau polymère peut seulement être appelé « semi-cristallin ».
Nucléation
Les premières cristallites se forment grâce aux mouvements thermiques des molécules avec des chaînes ou des portions de chaînes qui sont dans des positions favorables les unes par rapport aux autres. On parle de nucléation thermique ou homogène. Cependant, pour des raisons thermodynamiques, une croissance supplémentaire n'est possible que si les noyaux émergents dépassent une taille minimale critique. Sinon, les germes formés se désintègrent en raison de l'instabilité thermodynamique[3].
Cependant la nucléation due aux impuretés ou aux cristaux non fondus est plus fréquente que la nucléation thermique. Celle-ci est appelée « nucléation hétérogène ».
Les agents de nucléation spécialement ajoutés, les additifs et les charges peuvent grandement améliorer la formation de germes. Bien qu'il y ait beaucoup de travail sur le sujet des agents de nucléation, leur efficacité est mal comprise. Les agents de nucléation, qui ont une grande influence sur un type de polymère, sont inefficaces pour d'autres types de polymères. Beaucoup de bons agents de nucléation connus jusqu'à présent sont des sels métalliques d'acides organiques qui sont déjà présents sous forme cristalline aux températures de cristallisation du polymère.
Croissance
.svg.png.webp)
La croissance des cristaux se produit par l'assemblage d'autres segments de la chaîne polymère aux premières cristallites. Ceci a lieu dans une plage de température au-dessous de la température de fusion et au-dessus de la température de transition vitreuse. Si la température était trop élevée, les chaînes assemblées seraient de nouveau désassemblées par des mouvements thermiques. En dessous de la température de transition vitreuse, la mobilité des chaînes est trop faible et le mouvement des chaînes moléculaires est figé[4].
Entre les sections de chaîne parallèles agissent les forces intermoléculaires. Selon le type d'espèces atomiques impliquées, il peut s'agir d'interactions dipolaires ou de liaisons hydrogène. L'effet des forces dépend non seulement du type d'interaction mais également de la distance entre les sections de chaînes parallèles et détermine les propriétés mécaniques et thermiques du polymère[5].
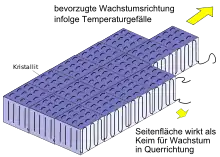
La croissance des régions cristallines se fait de préférence dans le sens du plus grand gradient de température (figure 3). Les surfaces latérales agissent également comme un germe pour la cristallisation. Cependant, le taux de croissance y est beaucoup plus faible. Des deux côtés des cristallites se trouvent les boucles de pliage amorphes, de sorte qu'aucune croissance ne peut avoir lieu dans ces deux directions. La croissance dirigée produit de longues bandes lamellaires de haute cristallinité qui poussent à partir de la graine et sont appelées « lamelles » (figure 3).
Les lamelles forment le bloc de construction de base de plus grandes superstructures cristallines. Avec des conditions de refroidissement statiques essentiellement isotropes, cela forme des sphérulites (figure 3) constituées de lamelles disposées radialement et symétriquement.
D'autre part, s'il y a un fort gradient de température dans l'échantillon, alors les lamelles sont disposées dans un arrangement en grande partie parallèle et donc dans une superstructure dirigée appelée « dendritique »[6]. De telles structures sont par exemple observées dans le polypropylène dans les zones de bord proches de la surface, lorsque la température du moule est choisie relativement froide lors du moulage par injection.
Dans les polymères à écoulement lent, des structures en forme d'haltère se forment lors du refroidissement, qui sont également décrites dans la littérature sous le nom de structures dites shish-kebab. La partie interne (noyau) est composée de chaines parallèles tandis que les bouts sont constitués de lamelles pliées.
Les composants qui se sont refroidis très rapidement (température de moule basse) ont eu trop peu de temps pour cristalliser complètement. Dans ce cas, il peut arriver plus tard (en partie aussi au cours des années) une post-cristallisation. Dans cette croissance cristalline secondaire, les propriétés des composants mécaniques changent. Puisque les chaînes sont plus denses à l'état cristallin, il y a aussi un retrait. Cela doit être pris en compte lors du procédé de moulage par injection[7].
Dans certains cas, les polymères sont stockés plus longtemps juste en dessous de la température de fusion de manière à augmenter la cristallinité. Ce procédé permet une orientation plus élevée des chaînes de polymère et empêche un retrait ultérieur pendant l'utilisation.
Cristallisation obtenue par étirement
La cristallisation pendant la solidification de la masse fondue est particulièrement importante dans le moulage par injection de composants en matière plastique. Le polymère peut habituellement être considéré comme une masse fondue relaxée au repos pendant le refroidissement.
.svg.png.webp)
D'autres conditions apparaissent lors de l'extrusion. Cette méthode est par exemple utilisée dans la production de fibres synthétiques ou artificielles et de films plastiques. Le polymère est pressé à travers une filière et les chaînes moléculaires sont légèrement préorientées.
En appliquant une contrainte de traction, l'orientation des chaînes moléculaires peut être significativement augmentée. Les fibres sont par exemple étirées à un multiple de leur longueur d'origine. Cet état correspond à une cristallisation partielle. La force de la fibre dans la direction longitudinale est considérablement augmentée. En règle générale, un traitement thermique ultérieur est effectué sous tension (thermofixage) pour obtenir un ordre plus élevé et pour réduire la tension, conduisant à une subséquente relaxation (rétrécissement). La fibre reste dimensionnellement stable. La forte anisotropie de la fibre est également mesurable grâce à ses propriétés optiques (biréfringence).
Une augmentation de la résistance par étirement ultérieur est également produite dans le processus de moulage par soufflage. Ici, une préforme, en PET par exemple, est gonflée avec de l'air comprimé à la taille et la forme prédéfinies. Les applications sont par exemple les réservoirs d'essence et les bouteilles en PET. Dans le même temps, la perméabilité aux gaz peut être considérablement réduite par l'étirement bidirectionnel[8].
L'étirement subséquent peut également être utilisé pour transformer des polymères amorphes en polymères semi-cristallins. Des structures cristallines lamellaires sont formées. Elles ne forment pas de superstructures sphérolitiques à la suite et restent ainsi complètement transparentes.
Cristallisation à partir d'une solution
Les polymères peuvent également être cristallisés à partir d'une solution ou par évaporation d'un solvant. Ce procédé dépend du degré de dilution : dans les solutions diluées, les chaînes moléculaires n'ont aucun lien entre elles et existent sous la forme de pelotes de polymère séparées dans la solution. L'augmentation de la concentration qui peut se produire via l'évaporation du solvant, induit une interaction entre les chaînes moléculaires et une éventuelle cristallisation[9]. La cristallisation de la solution peut entraîner des taux de cristallinité du polymère plus élevés. Par exemple, un polyéthylène hautement linéaire peut former des monocristaux de type plaquettaire avec une épaisseur de l'ordre de 10 à 20 nm lorsqu'il est cristallisé à partir d'une solution diluée. La forme cristalline peut être plus complexe pour d'autres polymères, y compris des pyramides creuses, des spirales et des structures dendritiques multicouches[10].
Un procédé très différent est la polymérisation par précipitation ; il utilise un solvant qui dissout les monomères individuels mais pas le polymère résultant. Lorsqu'un certain degré de polymérisation est atteint, le produit polymérisé et partiellement cristallisé précipite hors de la solution.
À l'aide de la résonance magnétique nucléaire à haute résolution, seule la partie dissoute d'une solution de polymère sursaturée est détectée. Ainsi, la diminution de la teneur dissoute pendant la cristallisation à partir de la solution et, par conséquent, la vitesse de cristallisation peuvent être déterminées[9].
Taux de cristallinité
Le taux de cristallinité désigne la fraction cristalline d'un polymère. Le taux de cristallinité peut être donné sous la forme d'une fraction massique, volumique ou molaire. Il dépend, entre autres, de l'histoire thermique du matériau.
Typiquement, des taux de cristallinité de 10 à 80 % sont possibles. L'obtention de taux de cristallinité plus élevés n'est possible qu'avec des molécules de faible masse moléculaire et / ou des échantillons spécialement recuits. Dans le premier cas, le matériau devient cassant, dans le second cas, le stockage prolongé à des températures juste inférieures à la température de fusion (recuit) signifie des coûts importants ce qui n'est utilisé que dans des cas particuliers. Les taux de cristallinité inférieurs à 10 % entraînent une tendance au fluage trop élevée si la température d'application du polymère est supérieure à sa température de transition vitreuse.
| Polymère | Taux typique de cristallisation (%)[2] | ρc (g/cm3) | ρa (g/cm3) |
|---|---|---|---|
| Polyamide (PA 6.6 et PA 6) | 35 - 45 | 1,24 | 1,08 |
| Polyoxyméthylène (POM homopolymère) | 90 | ||
| Polyoxyméthylène (POM copolymère) | 75 | ||
| Polytéréphtalate d'éthylène (PET) | 30 - 40 | 1,50 | 1,33 |
| Polytéréphtalate de butylène (PBT) | 40 - 50 | ||
| Polytétrafluoroéthylène (PTFE) | 60 - 80 | 2,35 | 2,00 |
| Polypropylène isotactique (PPi) | 70 - 80 | 0,95 | 0,85 |
| Polypropylène syndiotactique (PPs) | ≈ 30 - 40 | ||
| Polypropylène atactique (PPa) | ≈ 0 | – | – |
| Polyéthylène haute densité (PE-HD) | 70 - 80 | 1,0 | 0,85 |
| Polyéthylène basse densité (PE-LD) | 45 - 55 | 1,0 | 0,85 |
La plupart des évaluations des taux de cristallinité pour les thermoplastiques semi-cristallins supposent un modèle à deux phases avec des cristaux parfaits et des régions amorphes distinctes. Les écarts dus aux défauts, les zones de transition entre amorphe et cristallin sont susceptibles d'aller jusqu'à quelques pour cent.
Détermination
Les méthodes les plus couramment utilisées pour déterminer le taux de cristallinité dans les polymères sont la masse volumique, la calorimétrie différentielle à balayage (DSC), la diffraction des rayons X, la spectroscopie infrarouge et la résonance magnétique nucléaire (RMN). La valeur mesurée dépend de la méthode de mesure utilisée[11]. Par conséquent, il faut toujours indiquer la méthode utilisée en plus de la valeur du taux de cristallinité obtenue.
En plus des méthodes intégrales mentionnées ci-dessus, la distribution des régions cristallines et amorphes peut être visualisée par des techniques microscopiques, en particulier la microscopie en lumière polarisée et la microscopie électronique en transmission.
Mesure de la masse volumique
Les régions cristallines sont généralement plus denses que les régions amorphes. Il en résulte une masse volumique plus élevée qui varie typiquement jusqu'à environ 15 % selon le matériau (par exemple pour le polyamide 6 : ρc = 1,235 g/cm3 et ρa = 1,084 g/cm3). La masse volumique cristalline ρc est calculée à partir de la structure cristalline, tandis que la masse volumique amorphe ρa est mesurée expérimentalement sur un matériau amorphe. Le problème de la mesure de la masse volumique pour déterminer la cristallinité est que la masse volumique des régions amorphes dépend du refroidissement et peut être affectée par la présence d'humidité dans l'échantillon.
DSC
Lors de la fusion des polymères semi-cristallins, une énergie supplémentaire doit être utilisée pour convertir les structures cristallines solides en un état liquide amorphe. L'analyste parle ici d'un changement d'enthalpie endothermique. Le processus s'étend sur une grande plage de température. D'abord, les cristaux plus petits ou moins réguliers fondent. Lorsque la température augmente, des cristallites plus épais ou plus gros fondent jusqu'à ce que tout l'échantillon ait fondu[12]. L'enthalpie de fusion (énergie nécessaire pour faire fondre les cristaux) peut être déterminée par calorimétrie différentielle à balayage (DSC). En comparant avec une valeur de la littérature pour un matériau complètement cristallin (taux de cristallinité de 100 %), le taux de cristallinité calorimétrique de l'échantillon peut être calculé.
Diffraction des rayons X
Les distances atomiques produisent des signaux sous forme d'angles dans un diffractogramme. Dans les substances amorphes, des distances très différentes entre les chaînes moléculaires sont présentes. Cela conduit à une distribution très large dans le diagramme sous la forme d'une très grande courbe en cloche (halo). Les arrangements réguliers dans les zones cristallines, cependant, produisent des distributions beaucoup plus étroites sous la forme de pics. Dans les diffractogrammes de polymères réels, les halos et les pics sont superposés. En dépliant les pics, les intensités des pics et du halo peuvent être déterminées et à partir de cela, le taux de cristallinité par rayons X peut être calculé.
Spectroscopie infrarouge
Dans les spectres IR, des signaux supplémentaires (bandes) sont trouvés dans les polymères cristallins, qui sont absents dans les polymères amorphes de la même composition. Ces bandes proviennent des vibrations de déformation rendues possibles par l'arrangement régulier des chaînes moléculaires. À partir de l'évaluation de ces bandes, le taux de cristallinité infrarouge peut être calculé.
Résonance magnétique nucléaire (RMN)
Les régions cristallines et amorphes diffèrent dans la mobilité des protons. Cela montre des effets dans la forme de la ligne dans le spectre RMN. En prenant en compte le modèle structurel, des affirmations sur la cristallinité peuvent en être faites.
Facteurs influençant la cristallisation
La structure, la taille des cristaux et le taux de cristallinité dépendent d’un grand nombre de facteur.
Facteurs externes
Lors de la cristallisation pendant la solidification de la masse fondue, les facteurs externes qui influencent le taux de cristallisation d’un polymère sont la température de cristallisation et la vitesse de refroidissement. Dans le cas d'une cristallisation obtenue par étirement, ces facteurs sont la force d'étirage, la vitesse de déformation et le procédé de formation du film ou de la fibre de polymère.
Facteurs liés aux additifs et charges
Les agents de nucléation spécialement ajoutés, les additifs et les charges peuvent grandement affecter la cristallisation. Ils agissent comme des graines de nucléation et peuvent augmenter le taux de cristallisation.
Facteurs liés aux polymères
Une distribution étroite de la masse molaire, les masses molaires élevées, les chaînes polymères linéaires (non ramifiées), la tacticité, les forces intermoléculaires et la rigidité des chaînes principales augmentent le taux de cristallinité.
Les ramifications, les groupes latéraux volumineux et la réticulation diminuent le taux de cristallinité ou même le rendent très proche de zéro.
Tacticité
Plus l'ordre dans une macromolécule est grand, plus la probabilité de cristallisation de la molécule est grande.
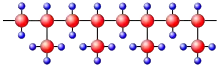
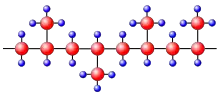
Dans le cas du polypropylène isotactique, les groupes latéraux CH3– sont régulièrement disposés d'un seul côté de la chaîne macromoléculaire (figure 5a). Il est ainsi possible que deux de ces parties de chaîne puissent s'appuyer les unes contre les autres presque dans toutes les positions. Dans le cas du polypropylène atactique (figure 5b), les groupes latéraux sont attachés des deux côtés de la chaîne de sorte qu'il n'y aura pas de juxtaposition de chaînes. La cristallisation est rendue alors beaucoup plus difficile ou même impossible. Les polymères atactiques cristallisent seulement lorsque les groupes latéraux sont très petits, comme dans le cas du polyfluorure de vinyle.
Forces intermoléculaires et rigidité des chaînes
Des forces intermoléculaires fortes et un squelette de chaîne rigide favorisent la formation de cristaux parce que les molécules préfèrent un arrangement ordonné avec une densité de compactage maximale pour maximiser le nombre de liaisons secondaires. Ainsi, les molécules ont tendance à s'organiser et à développer une structure cristalline. Un bon exemple est le Kevlar qui a un haut degré de cristallinité. Les groupes d'amides polaires dans le squelette sont fortement attirés les uns par les autres et forment de fortes liaisons hydrogène. Cela augmente la température de transition vitreuse et la température de fusion. Le fort taux de cristallinité et les fortes interactions intermoléculaires augmentent également considérablement la résistance mécanique. Les fibres de Kevlar sont parmi les fibres plastiques les plus résistantes du marché.
Taille des groupes latéraux
L'augmentation de la taille des groupes latéraux et du nombre de ramification rend plus difficile pour le polymère de se replier et de s'aligner le long de la direction de croissance des cristaux. Ainsi, les groupes latéraux encombrants et les ramifications réduisent la capacité et la probabilité de cristallisation d'un polymère. Par exemple, le polyéthylène ramifié présente un faible taux de cristallinité alors que le polyéthylène linéaire lui-même cristallise facilement.
Réticulation
La plupart des polymères réticulés (thermodurcissables) ne cristallisent pas parce que les sous-chaînes de polymères n'ont pas la liberté de se déplacer.
Propriétés des polymères semi-cristallins
Le comportement technique et les propriétés des plastiques sont essentiellement déterminés par la nature chimique des éléments de base, la longueur des chaînes mais aussi l'agencement des macromolécules[13].
La cristallisation des macromolécules modifie de manière significative les propriétés d'un matériau. Les propriétés d'un matériau semi-cristallin sont déterminées à la fois par les régions cristalline et amorphe du polymère. Cela montre une certaine connexion avec les matériaux composites, qui sont constitués de plusieurs substances. Les changements de propriété typiques avec une cristallisation croissante sont résumés dans le tableau adjacent et sont décrits plus en détail ci-dessous.
L'augmentation du taux de cristallinité augmente la masse volumique, la stabilité dimensionnelle, la rigidité, le module, la résistance à l'abrasion, la température de transition vitreuse, la température de fusion, la stabilité thermique et la résistance aux produits chimiques du polymère.
L'augmentation du taux de cristallinité diminue le gonflement, la résistance aux chocs, l'élasticité, l'allongement, la dilatation thermique, la perméabilité, la réactivité et la transparence du polymère.
Propriétés thermiques
En dessous de leur température de transition vitreuse, les polymères amorphes sont généralement rigides et fragiles en raison de la faible mobilité de leurs molécules. Si la température de transition vitreuse est dépassée, les chaînes moléculaires deviennent mobiles, ce qui donne des propriétés élastiques. Lorsque la température augmente, la mobilité des chaînes augmente et le matériau se ramollit. Le module d'élasticité diminue de manière significative. Une force constante appliquée à un polymère à des températures supérieures à la température de transition vitreuse entraîne une déformation viscoélastique, c'est-à-dire que le polymère commence à fluer[14]. La résistance à la chaleur est donc donnée pour les polymères amorphes juste en dessous de la température de transition vitreuse.
Des forces intermoléculaires relativement fortes dans les polymères semi-cristallins empêchent le ramollissement même au-dessus de la température de transition vitreuse. Leur module d'élasticité ne change significativement qu'à une température élevée (de fusion). Cela dépend aussi du taux de cristallinité : une cristallinité plus élevée donne un matériau plus dur et plus stable thermiquement, mais aussi plus fragile, alors que les régions amorphes procurent une certaine élasticité et résistance aux chocs.
Propriétés mécaniques
Les propriétés mécaniques du polymère résultent des propriétés des régions cristalline et amorphe. Plus la proportion de cristallites fortement tassées est élevée, plus le composant est dur, mais également plus il est fragile. Pour la production d'objets en plastique, une certaine cristallinité est tout à fait souhaitable car elle est responsable de la stabilité du plastique. Les zones amorphes, d'autre part, sont nécessaires pour conférer aux matériaux une certaine élasticité et résistance aux chocs.
Les plastiques sont des substances viscoélastiques, ce qui signifie que le comportement du matériau sous contrainte externe est fonction du temps. À charge constante, la déformation augmente avec le temps (fluage). Avec une déformation constante, la tension diminue avec le temps (relaxation).
Les matériaux étirés et les chaînes moléculaires alignés avec eux ne peuvent être que très peu étirés. Cet effet est observé avec les fibres synthétiques. Les nombreuses chaînes moléculaires s'étendant dans la direction de la traction se renforcent mutuellement et fournissent une augmentation significative de la résistance dans la direction des fibres.
La masse moléculaire (longueur de chaîne) a également une influence sur les propriétés du polymère. Avec l'augmentation de la longueur de la chaîne, les surfaces de contact augmentent, ce qui entraîne une augmentation de la résistance à la traction et une augmentation de la résistance chimique. En même temps, le nombre d'enchevêtrements augmente, ce qui améliore la ténacité à température ambiante, mais affecte négativement le comportement d'écoulement de la masse fondue. Lorsque les forces intermoléculaires deviennent plus fortes que la force de la chaîne, la résistance à la traction n'augmente plus malgré l'augmentation de la longueur de la chaîne. Une autre caractéristique des polymères semi-cristallins est une forte anisotropie de leurs propriétés mécaniques le long de la direction d'alignement moléculaire et perpendiculaire à celui-ci.
Masse volumique
.svg.png.webp)
Lors du refroidissement d'une matière plastique fondue, la mobilité des chaînes est réduite. Le volume de matériaux amorphes diminue de manière linéaire avec la température. En dessous de la température de transition vitreuse les chaînes sont immobiles. Le coefficient de dilatation thermique change, ce qui entraîne la pente de déviation de la courbe rouge de la figure 6.
Pour les matériaux cristallins, en dessous de la température de fusion on observe un agencement régulier des chaînes moléculaires et donc à une réduction importante de la distance entre les chaînes grâce à des forces intermoléculaires. Cela conduit à une augmentation de la masse volumique et à une réduction du volume spécifique (courbe bleu clair sur la figure 6).
Perméabilité
Le tassement plus dense des chaînes réduit le passage des gaz, conduisant à une réduction de la perméabilité, c'est-à-dire à une augmentation de l'étanchéité aux gaz.
Propriétés optiques
Les polymères cristallins sont généralement opaques en raison de la diffusion de la lumière sur les nombreuses frontières entre les régions cristallines et amorphes. L'opacité augmente avec la cristallinité, mais dépend aussi des différences des indices de réfraction. Le polypropylène syndiotactique est presque complètement transparent, tandis que le polypropylène isotactique avec une cristallinité comparable d'environ 50 % est très opaque. Cela peut s'expliquer par la structure cristalline différente entre ces deux polymères.
La coloration provient en grande partie de la phase amorphe. Les molécules de colorant peuvent mieux pénétrer entre les chaînes moléculaires du polymère. Les matériaux ayant un taux de cristallinité plus élevé peuvent donc être teintés moins bien que les matériaux ayant des zones plus amorphes[15].
Influence de la cristallisation sur les propriétés de traitement dans le moulage par injection
Lors du moulage par injection de thermoplastiques semi-cristallins, il faut tenir compte du fait que la chaleur supplémentaire libérée par le processus de cristallisation est dissipée, ce qui prolonge le temps de cycle. En outre, le plus grand changement de volume du matériau (dû au changement de densité pendant la cristallisation) doit être compensé par des temps de maintien plus longs[16].
Pour les polymères semi-cristallins, le retrait est également plus important que pour les polymères amorphes. Les conditions de refroidissement doivent être strictement respectées, car le processus de refroidissement a un effet durable sur le taux de cristallinité et donc sur les propriétés du matériau et du moulage. Bien qu'un refroidissement très rapide permette de supprimer en grande partie la cristallisation et de forcer une solidification presque amorphe, il faut du temps pour qu'une post-cristallisation ait lieu, ce qui signifie un rétrécissement et une distorsion qui vont avoir lieu plus tard.
Histoire des modèles de cristallisation
En 1925, Hermann Staudinger a découvert que certaines substances chimiques sont constituées de molécules à longue chaîne. Les études de diffraction des rayons X ont montré (en fonction du matériau) des spectres de diffraction typiques des cristaux. Des études plus détaillées ont révélé que certains polymères doivent être composés de nombreuses petites structures cristallines. La masse volumique du matériau a été calculée à partir des constantes de réseau obtenues à partir des études par rayons X et de la composition chimique connue. Cependant, la masse volumique calculée était toujours supérieure à la masse volumique déterminée expérimentalement.
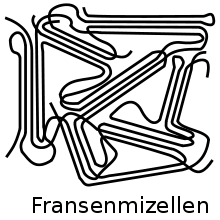
Par la suite, le modèle de «micelles frangées» a été développé par Abitz et Gerngroß (figure 7). Dans ce cas, les sections des chaînes moléculaires sont disposées parallèlement les unes par rapport aux autres sous forme de cristal. Les zones intermédiaires, cependant, sont amorphes. Une chaîne moléculaire, selon le concept, passe par différents cristaux. Après que les plus petits monocristaux de polymère aient été faits pour la première fois en 1957, on a trouvé que le modèle de la micelle frangées ne pouvait pas être maintenu pour la description des monocristaux. A. Keller postule en 1957 dans la revue Nature la formation de cristallites sous la forme de chaînes moléculaires pliées décrites ci-dessus (figure 2), qui vont d'un côté de la lamelle à l'autre et vice-versa[17].
Avec la méthode de contraste qu'il a développée en 1975, G. Kanig a non seulement visualisé la structure lamellaire du polyéthylène avec un microscope électronique, mais a également observé sa formation en refroidissant à partir de la masse fondue, ou sa fusion lorsque le matériau a été chauffé.
Notes et références
- Martin Bonnet, Kunststoffe in der Ingenieuranwendung: Eigenschaften, Verarbeitung und Praxiseinsatz polymerer Werkstoffe, Vieweg+Teubner Verlag, 2008 (ISBN 3-8348-0349-9) (Google Livres).
- Gottfried W. Ehrenstein, Polymer-Werkstoffe, Hanser Fachbuch, 1999 (ISBN 3-446-21161-6) (Google Livres).
- Georg Menges, Edmund Haberstroh, Walter Michaeli et Ernst Schmachtenberg, Werkstoffkunde Kunststoffe, Hanser Verlag, 2002 (ISBN 3-446-21257-4) (Google Livres).
- G.W. Becker, Ludwig Bottenbruch, Rudolf Binsack et D. Braun, Technische Thermoplaste. 4. Polyamide, Hanser Verlag, 1998 (ISBN 3-446-16486-3) (Google Livres).
- Wolfgang Weißbach, Werkstoffkunde und Werkstoffprüfung, Vieweg+Teubner Verlag, 2007 (ISBN 3-8348-0295-6) (Google Livres).
- (de) (en) Klaus Peter Großkurth, Kristallisation von Polypropylen, Institut für den Wissenschaftlichen Film, 1990, DOI 10.3203/IWF/C-1699 [vidéo].
- Wilbrand Woebcken, Klaus Stoeckhert et H. B. P. Gupta, Kunststoff-Lexikon, Hanser Verlag, 1998 (ISBN 3-446-17969-0) (Google Livres).
- Michael Thielen, Klaus Hartwig et Peter Gust, Blasformen von Kunststoffhohlkörpern, Hanser Verlag, 2006 (ISBN 3-446-22671-0), (Google Livres).
- J. Lehmann, « The observation of the crystallization of high polymer substances from the solution by nuclear magnetic resonance », Colloid & Polymer Science, vol. 212, no 2, , p. 167–168 (DOI 10.1007/BF01553085).
- Charles E. Carraher et Raymond Benedict Seymour (2003), Seymour/Carraher's polymer chemistry, CRC Press, p. 43–45 (ISBN 0-8247-0806-7).
- M. D. Lechner, K. Gehrke et E. H. Nordmeier, Makromolekulare Chemie, 4.
- Gottfried W. Ehrenstein, Gabriela Riedel et Pia Trawiel, Praxis der thermischen Analyse von Kunststoffen, Hanser Verlag, 2003 (ISBN 3-446-22340-1).
- Walter Michaeli, Einführung in die Kunststoffverarbeitung, Hanser Verlag, 2006 (ISBN 3-446-40580-1).
- Joachim Nentwig, Kunststofffolien, Hanser Verlag, 2006 (ISBN 3-446-40390-6).
- Burkhard Wulfhorst, Textile Fertigungsverfahren, Hanser Verlag, 1998 (ISBN 3-446-19187-9).
- Friedrich Johannaber et Walter Michaeli, Handbuch Spritzgießen, Hanser Verlag, 2004 (ISBN 3-446-22966-3).
- Hans K. Felger, Hermann Amrehn, Alexander von Bassewitz et Gerhard W. Becker, Polyvinylchlorid, Hanser Verlag, 1986 (ISBN 3-446-14416-1).