Syndrome du QT long
Le syndrome du QT long est un syndrome phénotypiquement et génétiquement hétérogène qui se caractérise par un allongement de l'intervalle QT sur l'électrocardiogramme de surface associé à un risque élevé de torsades de pointe ou de fibrillation ventriculaire pouvant entraîner syncope et mort subite.
| Symptômes | Trouble du rythme cardiaque |
|---|
| Médicament | Nadolol |
|---|---|
| Spécialité | Cardiologie |
| CIM-10 | I45.8 |
|---|---|
| CIM-9 | 426.82 |
| OMIM | 192500 |
| DiseasesDB | 11104 |
| eMedicine | 157826 |
| MeSH | D008133 |
![]() Mise en garde médicale
Mise en garde médicale
Génétique
Des mutations responsables de ce syndrome ont été décrites sur une dizaine de gènes, l'essentiel concernant LQT1 (ou KCNQ1), LQT2 (ou KCNH2) et LQT3 (ou SCN5A), chacun de ces derniers codant un canal ionique cardiaque[1]. Ces trois gènes concernent les trois quarts des syndromes et un cinquième n'ont pas de gène responsable identifié[2]. Certains syndromes sont de type autosomique dominant, d'autres sont de type autosomique récessif.
Des mutations sur d'autres gènes ont été décrites, responsables de formes atypiques de la maladie, dont le TRDN[3], le CALM1 (en), CALM2 (en), et CALM3 (en)[4].
D'autres gènes sont indirectement impliqués ou avec une pénétrance faible, dont le KCNE1 (en)[5].
Épidémiologie
Une mutation est présente environ chez une personne sur 2000[6]. À la naissance, un bébé sur 2 000 aurait cette anomalie électrocardiographique[7].
Description
Il peut se présenter de manière isolée ou sous forme d'un syndrome associant plusieurs malformations :
- le syndrome de Romano-Ward ;
- le syndrome de Jervell-Lange-Nielsen, associé à une atteinte de l'audition ;
- le syndrome d’Andersen-Tawil ;
- le syndrome de Timothy.
Le syndrome peut être découvert à l'occasion d'un malaise, voire d'une syncope. Sa première manifestation peut être celle d'une mort subite. Il est rare que la première manifestation se fasse après l'âge de 40 ans.
Il peut être également découvert lors d'un électrocardiogramme fait pour une raison autre, ou lors d'une enquête familiale lorsque l'un des membres de la famille est porteur de ce syndrome.
Diagnostic
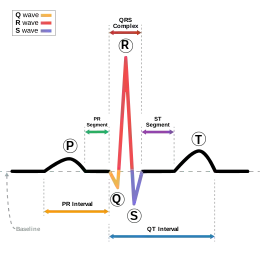
Il repose sur la mesure de l'intervalle QT sur l'électrocardiogramme : durée entre le début de l'onde QRS, synchrone de la dépolarisation des ventricules, et la fin de l'onde T, correspondant à la repolarisation des ventricules.
La probabilité est forte si la mesure de l'espace QT suivant la formule de Bazett : QT corrigé = QT/racine carrée de l'espace RR précédent est supérieur à 440 millisecondes. Il faut, naturellement, éliminer les autres causes de prolongation du QT : prises de certains médicaments, hypokaliémie (taux bas en potassium sanguin). Le QT peut être faiblement prolongé, rendant le diagnostic plus difficile. Le « score de Schwartz », reposant sur différents critères dont l'aspect de l'ECG, les antécédents de syncopes et les antécédents familiaux, peut aider dans ces cas[8]. Le diagnostic peut également être aidé par la mesure de l'intervalle QT quelques minutes après une épreuve d'effort[9].
Le risque de faire une syncope ou une mort subite semble proportionnel avec la durée du QT[10]. Il est également plus important en cas d'antécédent de syncope[11].
Le typage de l'anomalie génétique est intéressant dans un but pronostic, certaines mutations étant plus graves que d'autres. De même, certaines mutations peuvent présenter des tableaux particuliers : une atteinte du gène LQT1 donne typiquement des syncopes à l'effort[12] répondant particulièrement bien aux médicaments de type bêta-bloquants et avec très peu de morts subites sous traitement. Au contraire, une mutation du gène LQT2 entraîne des syncopes plutôt aux émotions et plus rarement à l'effort, avec une efficacité moindre des bêta-bloquants et un risque de morts subites nettement plus important[13], les porteurs d'une mutation sur le gène LQT3 ayant un pronostic comparable. Le typage génétique a permis de découvrir des formes à « QT normal » de la maladie, témoignant d'une pénétrance faible[14].
Traitement
Des recommandations américaines et européennes, concernant les troubles du rythme d'origine génétique, ont été publiées en 2013[15].
Les médicaments bêta-bloquants ne diminuent pas le QT mais abaissent le risque de complications[16]. Ils seraient plus efficaces chez les porteurs d'une mutation sur le gène LQT1[17]. Le propranolol et le nadolol seraient plus efficaces que le métoprolol pour la prévention d'accidents cardiaques[18]. En particulier, le nadolol semble mieux marcher dans les atteintes du LQT2[19]. La mexilétine (en) raccourcit le QT des LQT3 en diminuant très sensiblement le risque de survenue d'un événement rythmique grave[20]. Cependant, le corgard reste l'un des médicaments les plus fiables concernant cette maladie.
Chez les patients jugés à haut risque de complication, la pose d'un défibrillateur implantable doit être proposée[21].
Les sports de compétition sont interdits (exceptions possibles : golf, tir à l'arc, billard) et les activités aquatiques (natation, baignades, etc.) sont à éviter. En revanche le sport occasionnel ou « de loisir » est autorisé. Il est préférable d'en discuter avec son cardiologue qui donnera des indications au cas par cas[22].
Le stress auditif (sonnerie stridente par exemple) est à éviter pour les personnes atteintes du syndrome du QT long de type 2. En effet, ces personnes sont plus sensibles à une brusque accélération du rythme cardiaque (qui peut être causé entre autres par un coup de stress).
Notes et références
- (en) Roden D, « Long-QT Syndrome » N Eng J Med. 2008;358:169-176
- (en) Schwartz PJ, Ackerman MJ, George AL, Wilde AA, « Impact of genetics on the clinical management of channelopathies » J Am Coll Cardiol. 2013;62:169-180.
- Altmann HM, Tester DJ, Will ML et al. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive Llong-QT syndrome and pediatric sudden cardiac arrest: Elucidation of the triadin knockout syndrome, Circulation, 2015;131:2051-2060
- Adler A, Novelli V, Amin AS et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome, Circulation, 2020;141:418–428
- Roberts JD, Asaki SY, Mazzanti A et al. An international multicenter evaluation of type 5 long QT syndrome: a low penetrant primary arrhythmic condition, Circulation, 2020;141:429–439
- (en) Schwartz PJ, Priori SG, Napolitano C, « How really rare are rare diseases? The intriguing case of independent compound mutations in the long QT syndrome » J Cardiovasc Electrophysiol. 2003;14:1120-1121
- (en) Schwartz PJ, Stramba-Badiale M, Crotti L et al. « Prevalence of the congenital Long-QT Syndrome » Circulation 2009;120:1761-1767
- (en) Schwartz PJ, Moss AJ, Vincent GM, Crampton RS, « Diagnostic criteria for the long QT syndrome: an update » Circulation 1993;88:782–784
- (en) Sy RW, van der Werf C, Chatta IS et al. « Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands » Circulation 2011;124:2187–2194
- (en) Priori SG, Schwartz PJ, Napolitano C et al. « Risk stratification in the long-QT syndrome » N Engl J Med. 2003;348:1866-1874
- (en) Moss AJ, Schwartz PJ, Crampton RS et al. « The long QT syndrome: prospective longitudinal study of 328 families » Circulation 1991;84:1136–1144
- (en) Ruan Y, Liu N, Napolitano C, Priori SG, « Therapeutic strategies for long-QT syndrome: Does the molecular substrate matter? » Circ Arrhythmia Electrophysiol. 2008;1:290-297
- (en) Schwartz PJ, Priori SG, Spazzolini C et al. « Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias » Circulation 2001;103:89–95
- (en) Priori SG, Napolitano C, Schwartz PJ, « Low penetrance in the long-QT syndrome: clinical impact » Circulation 1999;99:529–533
- SG Priori, AA Wilde, M Horie et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes, Heart Rhythm, 2013;10:1932-63
- (en) Sauer AJ, Moss AJ, McNitt S et al. « Long QT syndrome in adults » J Am Coll Cardiol. 2007;49:329-337
- (en) Priori SG, Napolitano C, Schwartz PJ et al. « Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers » JAMA 2004;292:1341-1344
- (en) Chockalingam P, Crotti L, Girardengo G et al. « Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: higher recurrence of events under metoprolol » J Am Coll Cardiol. 2012;60:2092-2099
- (en) Abu-Zeitone A, Peterson DR, Polonsky B, McNitt S, Moss AJ, « Efficacy of different beta-blockers in the treatment of long QT syndrome » JACC 2014;64:1352–1358
- Mazzanti A, Maragna R, Faragli A et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3, J Am Coll Cardiol, 2016;67:1053–1058
- (en) Zipes DP, Camm AJ, Borggrefe M et al. « ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death » Europace 2006;8:746–837
- Vincent Probst - cardiologie et maladies vasculaires « Le syndrome du QT long - sports contre-indiqués » CHU de Nantes, 2012