Syndrome IPEX
Le syndrome d'immunodérégulation, de polyendocrinopathie et d'entéropathie lié au chromosome X appelé syndrome IPEX est une maladie rare associant un déficit immunitaire, une polyendocrinopathie et une entéropathie. C'est une affection récessive liée à une mutation du gène FOXP3 situé sur le chromosome X. Cette mutation provoque un défaut de régulation de la réponse immunitaire induisant des atteintes auto-immunes, des réactions allergiques dues à l'absence d'une population de lymphocytes : les lymphocytes T régulateurs.
Historique
En 1949, le caractère « Scurfy » est apparu spontanément au sein d'une lignée murine. Il s'agit d'un caractère transmis de façon récessive lié au chromosome X. Les souris montrent des symptômes proches de ceux de l'IPEX comme des atteintes cutanées et digestives, et meurent de façon précoce aux alentours du vingt-quatrième jours de vie. Les souris « Scurfy » constituent ainsi un modèle animal de la maladie[1].
En 1982, Powell et al.[2] ont fait des études génétiques sur des familles atteintes par le syndrome. Ils ont ainsi montré que cette maladie est liée au chromosome X.
En 2001, des études de liaison dans les familles atteintes et les travaux chez la souris scurfy ont abouti à l'identification du locus foxp3 sur le chromosome X[3].
Population touchée
Cette maladie se manifeste toujours chez le jeune garçon. Elle apparaît le plus souvent au cours du premier mois de vie et peut être très rapidement fatale pour le nourrisson. Les enfants dont la maladie ne s'est pas développée dès la naissance ont une espérance de vie qui dépasse rarement 8 ans.
Symptômes
Le syndrome IPEX provoque les symptômes[4] suivants :
- Une diarrhée profuse, liquidienne, sécrétoire et rarement glairo-sanglante, causant un retard important de croissance staturo-pondérale.
- Une dermatite de type atopique et diffuse, pouvant être de type psoriasis, ichtyose ou eczéma[5].
- Un diabète insulinodépendant de type I.
- Apparition de complications infectieuses ou virales.
Ces symptômes sont souvent associés à des signes d'auto-immunité comme des anémies hémolytiques, des troubles hydroélectriques, des hyper ou hypothyroïdies, une atteinte rénale, une alopécie[5]. Tous ces symptômes sont liés à une présence anormale d'auto-anticorps dans l'organisme. Cette maladie est proche d'autres syndromes atteignant le système immunitaire comme le syndrome de Wiskott-Aldrich.
Diagnostic
Le diagnostic est effectué à partir du tableau clinique observé chez l'enfant. La découverte néonatale de diabète de type I, de diarrhées graves couplées à l'apparition d'eczéma, de signes d'auto-immunité (présence d'auto-anticorps notamment contre l'harmonine et la villine[6], deux protéines exprimées dans le rein et le tube digestif, ce qui pourrait en expliquer les symptômes) entraînera un bilan immunologique et une recherche de mutation du gène FOXP3. D'autres examens comme une coloscopie accompagnée de biopsies, une recherche de diabète de type I, un dosage des IgE en plus du bilan immunitaire standard, permettront de confirmer le diagnostic.
Traitements et perspectives
- Traitements des symptômes :
- un traitement endocrinien (insulinothérapie par exemple)
- des transfusions sanguines
- une rééquilibration hydroélectrolytique intaveineuse pour compenser les pertes diarrhéiques
- une alimentation parentérale exclusive
une immunosuppression médicamenteuse pour diminuer les réactions auto-immunes et allergiques. Ce traitement présente une toxicité à long terme et l'augmentation d'infections opportunistes ainsi que des risques de récidive de la maladie[7].
La greffe de moelle osseuse permet d'apporter des cellules souches saines capables de se différencier en lymphocytes T régulateurs fonctionnels et donc de restaurer, en théorie définitivement, la régulation normale des réponses immunitaires[8]. Elle constitue la meilleure option thérapeutique chez les patients ayant peu d'atteinte des organes[9], même si les effets secondaires liés à la prise d'immunosuppresseurs sont non négligeables (toxicité, sensibilité aux infections, maladie du greffon contre l'hôte,...).
- Perspective : la thérapie génique
Elle viserait à substituer le gène foxp3 défectueux du patient par un gène foxp3 fonctionnel au sein des cellules souches hématopoïétiques.
Mécanismes moléculaires à l'origine de la maladie
Gène foxp3
Près de 70 mutations responsable du syndrome ont été décrits[5].
Fonction de la protéine FoxP3
La protéine FoxP3 est un facteur de transcription, elle est capable de s'associer au principal facteur de transcription de l'activation lymphocytaire NFAT pour modifier l'expression génique et inhiber la transcription des gènes d'activations des lymphocytes T auxiliaires.
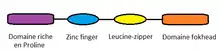
Elle[10] est constituée de plusieurs domaines :
le domaine riche en proline et le domaine en doigt de zinc interagissent avec d'autres facteurs de transcriptions (NFAT,...). Le domaine en glissière à leucine permet l'homo- ou l'hétérodimérisation. Le domaine forkhead se lie à l'ADN ce qui déclenche des signaux de localisation intra-nucléaire.
Le facteur de transcription FoxP3 est essentiel au développement et au fonctionnement d'une sous-population de lymphocytes T : les lymphocytes T régulateurs dont le rôle est de maintenir la tolérance immunologique aux antigènes du soi et aux antigènes non dangereux.
Dysfonctionnements
Les patients atteints du syndrome IPEX présentent des mutations de foxp3 entraînant des modifications sur les domaines forkhead[11], glissière à leucine ou riche en proline, induisant des dysfonctionnements plus ou moins graves. La mutation du gène foxp3 n'est pas la seule cause du syndrome IPEX. En effet d'autres gènes impliqués dans le développement ou le fonctionnement des lymphocytes T régulateurs peuvent causer ce syndrome (ex : mutations autosomiques récessives de l'antigène de surface CD25).
Rôle
Les lymphocytes T régulateurs sont directement impliqués dans la suppression des réponses immunitaires. Leur rôle principal est de maintenir la tolérance en empêchant l'activation des lymphocytes T, notamment des lymphocytes T auto-réactifs. En effet, ces lymphocytes T reconnaissent des antigènes du soi et peuvent en cas d'activation provoquer des maladies auto-immunes. Les lymphocytes T régulateurs empêchent également l'activation des lymphocytes T spécifiques d'antigènes étrangers non dangereux susceptibles de provoquer des réactions d'hypersensibilités.
Dysfonctionnements
Chez les patients atteints du syndrome, les lymphocytes T régulateurs sont non fonctionnels ce qui déclenche une suractivation du système immunitaire provoquant l'apparition de symptômes (auto-immunité, réactions d'hypersensibilités,...). Le taux de lymphocytes T exprimant FoxP3 n'est pas forcément diminué chez ces patients mais ils subissent des altérations fonctionnelles dans leur capacité à inhiber les lymphocytes T activés, on observe donc une augmentation anormale des lymphocytes T CD4+ activés dans la circulation, ce qui provoque une absence de tolérance aux antigènes non dangereux et aux antigènes du soi.
Notes et références
- Wildin RS, Ramsdell F, Peake J et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy, Nat Genet, 2001;27:18-20
- Powell BR, Buist NR, Stenzel P, An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy, J Pediatr, 1982;100:731-737
- Bennett CL, Christie J, Ramsdell F et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3, Nat Genet, 2001;27:20-21
- (it) Toona Prod, Febz , Bizza., « Ipex syndrome consortium », sur ipexconsortium.org (consulté le ).
- Husebye ES, Anderson MS, Kämpe O, Autoimmune polyendocrine syndromes, N Eng J Med, 2018;378:1132-1141
- Lampasona V, Passerini L, Barzaghi F et al. Autoantibodies to harmonin and villin are diagnostic markers in children with IPEX syndrome, PLoS One, 2013;8:e78664-e78664
- Barzaghi F, Amaya Hernandez LC, Neven B et al. Long-term follow up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study, J Allergy Clin Immunol, 2018;141:1036-1049
- Wildin RS, Smyk-Pearson S, Filipovich AH, Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome, J Med Genet, 2002;39:537-545.
- Barzaghi F, Amaya Hernandez LC, Neven B et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study, J Allergy Clin Immunol, 2018;141:1036-1049.e5
- A. Marabelle et al., « De l’Ipex à foxp3 : une nouvelle contribution de la pédiatrie à la compréhension du système immunitaire », Arch. pédiatr., vol. 15, no 1, , p. 55-63
- Brunkow ME, Jeffery EW, Hjerrild KA et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse, Nat Genet, 2001;27:68-73
Lien externe
- (en) « IPEX syndrome consorium » (consulté le )