Plasticité et endommagement d'un polymère
Les polymères sont des matériaux complexes dont le comportement face à la pression varie : sous de faibles contraintes, ils réagissent en se déformant de manière élastique et viscoélastique ; sinon, ils suivent un comportement plastique dont il résulte ce qu'on appelle de l'endommagement. Au niveau macroscopique, les phénomènes de déformation plastique se traduisent principalement par l’apparition de bandes de cisaillement et/ou de craquelures.
La plasticité est influencée par divers paramètres que sont principalement la température et la vitesse de sollicitation. Elle n’est pas forcément néfaste pour les polymères, cependant au-delà du seuil de plasticité (défini par les échelles de Tresca ou de Von Mises), l’endommagement nuit aux propriétés mécaniques (rigidité, résistance mécanique) et peut mener à la rupture de la pièce.
Courbe contrainte-déformation
Pour comprendre le comportement d'un matériau polymère lors d'une sollicitation en traction par exemple, et donc prédire sa rupture, les polyméristes s'appuient sur des courbes décrivant l'évolution de la contrainte en fonction de la déformation. Ces courbes, décrivant le comportement mécanique, sont découpées en différents domaines.

Pour des petites déformations, le polymère suit un comportement linéaire décrit par la loi de Hooke :
- .

C'est le domaine élastique.
Pour des déformations légèrement supérieures, on entre dans le domaine viscoélastique.
Le seuil de plasticité, noté y, représente la limite entre le domaine élastique et le domaine plastique. Typiquement, ce seuil intervient aux environs de 5 à 15 % de déformation.
Pour le domaine plastique, on distingue différentes zones :
- l'apparition de la striction, où la déformation plastique est instable du fait de la localisation des déformations. Cette striction est générée par les bandes de cisaillement ;
- la zone d'étirage, qui s'étend sur de grandes déformations et qui correspond à l'allongement et à l'alignement des chaînes de polymère à contrainte constante ;
- le durcissement final, étape durant laquelle les chaînes sont en extension ;
- la rupture, où le polymère cède.
Observations macroscopiques de l’endommagement
Les bandes de cisaillement
Lorsque l’on déforme un matériau polymère par cisaillement, ce dernier peut réagir de deux manières différentes. La déformation se répartit soit de manière homogène, comme c’est le cas avec la plupart des polymères thermoplastiques, soit elle est fortement localisée et visible sous forme de fines bandes, appelées bandes de cisaillement (shear band en anglais). Leur épaisseur varie de quelques micromètres à un millimètre.
On remarque dans la plupart des cas que les bandes de cisaillement sont inclinées par rapport à la direction de traction d’un angle de 45° environ, car c’est dans ce plan que la contrainte de cisaillement est maximale (Facteur de Schmid (en)).
Le type de polymère conditionne à la fois l’apparition ou non de bandes de cisaillement, et la vitesse de déformation et la température à laquelle la déformation a lieu. Ainsi, pour des températures inférieures à 0,8.Tg, on a un comportement homogène ; au-delà, les déformations deviennent localisées.
Ce mécanisme d’endommagement est souvent plus faible pour des essais de traction. En effet, les craquelures sont alors prédominantes. À l’inverse, pour un essai de compression, les bandes de cisaillement l’emportent.
L’apparition des bandes de cisaillement s’accompagne d’un adoucissement, c’est-à-dire d’un ramollissement du matériau, ce qui se traduit par une chute de la contrainte. Ensuite, plus la contrainte augmente et plus le nombre de bandes de cisaillement augmente, et donc la déformation du matériau.
Les craquelures
Les craquelures (craze en anglais) définissent la tenue à la rupture des polymères. La formation de craquelures est appelée craquelage (crazing en anglais).
L’hypothèse isovolumique habituellement pratiquée dans les études de déformation des matériaux (comme les métaux) n’est pas valable pour les polymères. En effet, lors d’un essai de traction sur ces derniers, du vide se forme au sein des chaînes, d'où une augmentation du volume total.
Une craquelure est constituée majoritairement de vacuoles (zones de vide pouvant atteindre 30 à 40 % du volume total) et autour desquelles on observe des fibrilles (chaînes macromoléculaires orientées). Ces dernières ont un comportement mécanique particulier expliquant les phénomènes de rupture du matériau. On peut observer les craquelures par microscopie électronique à balayage.
Les craquelures sont le fruit d’un processus de transformation d’énergie. En effet, les deux surfaces de part et d’autre de la craquelure emmagasinent des énergies de surface de rupture ; énergie auparavant stockée sous forme élastique dans le matériau. Donc les craquelures sont bénéfiques car elles absorbent l’énergie mécanique, renforçant ainsi le polymère et évitant parfois la rupture.
La nucléation
L’approche de la nucléation des craquelures est problématique. En effet, sur le plan physique, on constate une discontinuité des propriétés mécaniques (telle que la densité), ce qui pose problème. Il s’agit néanmoins d’un mécanisme de cavitation.
La nucléation a toujours lieu dans une zone affaiblie, c’est-à-dire où il y a des impuretés, une densité d’enchevêtrements plus faible ou un défaut géométrique (fissure). Il s’agit donc souvent d’une zone de concentration de contraintes.
Remarquons que pour les alliages de polymères, comme le polystyrène choc, le phénomène de craquelure est important compte tenu du nombre élevé de sites de nucléation. On remarque d’autre part, qu’expérimentalement la nucléation est plus faible pour des polymères fortement orientés ; pour les résines thermodurcissables, le réseau est tellement réticulé que les craquelures ne peuvent pas apparaître.
Les craquelures sont également impossibles si l'on exerce une compression, car alors la pression exercée empêche la création de vide. Enfin, ce phénomène ne se produit que pour des températures de l’ordre de 0,8.Tg.
Les craquelures extrinsèques naissent localement dans les zones affaiblies, parfois avant le seuil de plasticité ; les craquelures intrinsèques apparaissent dans le volume tout entier pour des températures très proches de la température de transition vitreuse (Tg).
Numériquement, on peut établir une valeur limite d’apparition des craquelures.
La croissance
La croissance des craquelures s’explique plus facilement par deux mécanismes régissant l’allongement des fibrilles sous contrainte. Le premier est un mécanisme de fluage qui entraîne la croissance des craquelures perpendiculairement à la direction d’étirement. Le deuxième repose sur l’approvisionnement de la matière située dans la masse totale du polymère.
Typiquement, l’allongement peut atteindre des valeurs de 500 %, le diamètre des fibrilles restant constant (10 nm). Pour le taux de vide dans une craquelure, cela s’élève à 50 voire 80 %.
Le mécanisme d’extraction de matière est prépondérant pour la croissance des craquelures, cependant le mécanisme minoritaire de fluage a une importance capitale dans la rupture finale du matériau.
D’un point de vue mécanique, un polymère infesté de craquelures n’est pas ruiné ; au contraire, la présence des fibrilles permet aux craquelures de soutenir une charge dans la direction perpendiculaire à leurs faces.
Craquelures et rupture
La présence de craquelures au sein d’un matériau polymère n’est pas néfaste comme nous venons de le voir. Cependant, si les fibrilles des craquelures se rompent, le polymère se dirige tout droit vers sa ruine.
La rupture des fibrilles est liée au désenchevêtrement puis à la rupture des chaînes de polymères. Suivant la température, on distingue deux modes de rupture :
- à basse température (T < Ttransition secondaire), la rupture se produit à n’importe quel endroit de la fibrille ;
- à haute température (T > Ttransition secondaire), la rupture a lieu au milieu des fibrilles.
La craquelure devient alors une microfissure qui croît jusqu’à la rupture.
Compétition entre bandes de cisaillement et craquelures
Suivant le type d’essai réalisé sur l’échantillon, on observe en traction majoritairement des craquelures et en compression des bandes de cisaillement.
Lors d’un essai de compression, les craquelures ne peuvent se former ; ce sont donc les bandes de cisaillement qui l’emportent et qui se développent. A contrario, en traction, les craquelures apparaissent pour des valeurs de déformations inférieures à 10 % ; ce qui limite le développement des bandes de cisaillement.
Le polystyrène est un bel exemple de cette compétition. En effet, il a un comportement fragile en traction (lié à la présence de crazes se transformant en fissures et amenant à la rupture) alors qu’il est ductile en compression (car les bandes de cisaillement entraînent un ramollissement du matériau).
On peut également assister à une osmose entre les phénomènes. En effet, des craquelures peuvent générer des bandes de cisaillement.
Carte de résistance mécanique
Devant la diversité et l’interaction des phénomènes mis en jeu lors d’une déformation, il est très utile, pour un ingénieur, de pouvoir utiliser une « carte » afin de prévoir le comportement d’un polymère pour une sollicitation donnée, à une température donnée. On peut déterminer alors facilement comment se comportera le matériau polymère après sa mise en œuvre, par exemple.
On peut trouver des exemples de carte de résistance comme pour le PMMA. Elle est généralisable à tous les polymères thermoplastiques amorphes.
Toutefois, cette carte n’est pas universelle et des mécanismes de rupture prématurée peuvent avoir lieu en présence de défauts indésirables, par exemple. Il faut donc l’utiliser avec parcimonie.
Apparition de la plasticité : seuil et critères de plasticité
La définition de la contrainte de glissement au seuil de plasticité est générale et valable pour la majorité des matériaux. Comme nous l’avons évoqué précédemment, en dehors du domaine élastique, les déformations dues à la présence des plans de glissement deviennent irréversibles.
Pour mieux détailler l’aspect mécanique des déformations, on considérera un essai de traction uniaxial.
La force F, faisant un angle α avec la normale au plan de glissement et un angle θ avec la direction de glissement, alors sa composante dans la direction de glissement est F*cos(θ).
Ainsi, la contrainte exercée sur la surface A du plan, appelée contrainte de cisaillement ou de glissement, ou encore scission s’écrit :
- σ =(F/A)*cos(θ)*cos(α) = σtraction*cos(θ)*cos(α) (3.1)
Seuil de plasticité
Si on augmente la force F à partir de zéro, la déformation plastique du cristal commence à se produire pour une valeur maximale de σc (scission critique de glissement). En général, on peut montrer que le début de la plasticité a lieu lorsque le seuil de plasticité vaut :
- σy = 2*σc (3.2)
Certains polymères ne présentent pas de seuil de plasticité σy. C’est le cas des polymères fragiles où l’échantillon casse au stade viscoélastique, ou des élastomères dont le comportement est hyperélastique. Si ce seuil de plasticité σy n’existe pas, on montre que la déformation plastique débute pour des valeurs de l’ordre de 5 à 10 %.
Critères de plasticité généraux
On considère le cas d’une déformation uniaxiale (traction-compression), ce qui permet de définir un seuil de plasticité σy en traction ou compression.
On établit alors un critère de plasticité attaché à une notion de valeur critique, à partir de laquelle se produit une déformation irréversible.
Deux des plus courants critères sont détaillés ici :
Pour simplifier les choses, on choisit les trois axes principaux orthogonaux, tels que les contraintes de cisaillement soient nulles. Le tenseur des contraintes devient :
- (3.3)

Le critère de Tresca

Le critère de Tresca, établi en 1867, spécifie qu'un matériau va se déformer plastiquement lorsque la contrainte de cisaillement maximale atteint une valeur critique σc. Si les trois contraintes principales sont telles que σ1 >σ2 >σ3, le critère de Tresca s’écrit :
- (σ1-σ3)=2*σc=σy (3.4)
Le critère de Von Mises
Le critère de Von Mises, établi en 1913, prédit que la déformation plastique débute lorsque l’énergie de distorsion atteint une valeur critique ; il peut s’écrire par une relation entre les contraintes principales :
- (σ1-σ2)² + (σ2-σ3)² + (σ3-σ1)² = 2*σy² (3.5)
Représentation graphique des critères de Tresca et de Von Mises
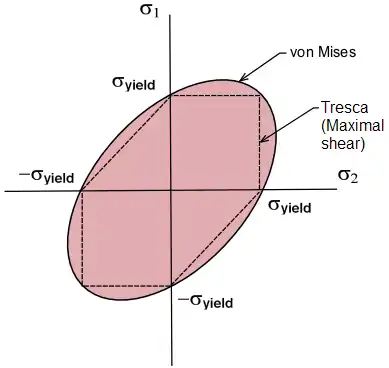
Pour simplifier les calculs des deux critères, on annule la contrainte σ3. Le critère de Von Mises devient donc :
- (σ1/σy)² + (σ2/σy)² - (σ1/σy)*(σ2/σy) = 1 (3.6)
Ceci est l’équation d’une ellipse.
De même, le critère de Tresca se traduit par six segments de droites :
- Si σ1>σ2>0 → σ1=σy
- Si σ1>0>σ2 → σ1-σ2=σy
- Si 0>σ1>σ2 → σ2=-σy
- Si σ2>σ1>0 → σ2=σy
- Si σ2>0>σ1 → σ2-σ1=σy
- Si 0>σ2>σ1 → σ1=-σy
On peut ainsi tracer l’ensemble de ces courbes sur un même graphique :
sur cette figure, on remarque que les deux critères sont très proches et que les deux courbes coïncident pour chaque sommet de l’hexagone de Tresca, c’est-à-dire que les deux critères prédisent un seuil de plasticité identique en traction comme en compression. Ce constat est observé en général pour tous les matériaux hormis pour les polymères car ces derniers sont sensibles à la pression hydrostatique.
Effet de la pression hydrostatique sur le seuil de plasticité
Expérimentalement, on constate pour la plupart des polymères que :
- le seuil de plasticité σy en traction est plus faible que celui en compression uniaxiale. En effet, la contrainte de compression augmente fortement entre les unités constitutives du polymère et donc le glissement va être plus difficile ;
- le seuil de plasticité σy croît fortement avec la pression hydrostatique car les polymères sont sensibles à la pression, du fait de l’existence du volume libre disponible entre les unités constitutives.
Les critères de Von Mises et de Tresca ne tenant pas compte de ces deux effets, nous allons désormais les modifier pour prendre en compte l’influence de pression sur le seuil de plasticité.
Critère de Tresca modifié
Pour rendre compte de l’effet hydrostatique dans le critère de Tresca, la valeur critique σc devient une fonction linéaire de la pression hydrostatique, notée P :
- σc=σc0 - µ*P (3.7)
où :
- µ est le coefficient de frottement dépendant du matériau. En général pour les polymères, ce coefficient µ est positif ;
- σc0 est la valeur de σc lorsqu'aucune pression n’est appliquée ;
- P est la moyenne des trois contraintes principales : P=(σ1+σ2+σ3)/3.
Remarquons que P en tension est positif et P en compression est négatif.
Cette formule indique que la valeur critique σyc augmente en compression (la pression P étant négative en compression), et σyt diminue en tension (pression positive). La plasticité en compression est donc plus difficile qu’en traction.
Critère de Von Mises modifié
De même que le critère de Tresca, le critère de Von Mises peut se transformer en introduisant la pression hydrostatique P. P étant toujours défini comme la moyenne des trois contraintes principales. Pour la plupart des polymères solides, le critère de Von Mises modifié se formule par :

où A et B sont deux constantes dépendant de la pression hydrostatique.
On peut déterminer A et B en mesurant les seuils de plasticité en traction uniaxiale et en compression uniaxiale , car dans ces deux cas et .




Ainsi, le critère de Von Mises prend la forme suivante :

Représentation graphique des critères de Tresca et Von Mises modifiés
Graphiquement, le critère de Von Mises modifié s'apparente à une ellipse déformée au sein de laquelle s'inscrit toujours le critère de Tresca.
Les critères classiques prédisaient un seuil de plasticité identique en traction ou en compression, or il s’avère que les critères modifiés imposent un seuil de plasticité supérieur en compression qu’en traction.
Par analogie avec les critères de plasticité de Tresca et Von Mises, on peut établir un critère d’endommagement par craquelures étant donné que les craquelures apparaissent au-delà d’une déformation critique fonction de la pression hydrostatique. On peut ensuite relier cette déformation à la contrainte par la loi de Hooke (1.1). On montre alors que :
- σ1-ν*σ2 ≥ C + D/(σ1+σ2) (3.10)
avec ν le coefficient de Poisson et C et D deux constantes fonction du temps et de la température.
La représentation graphique subit par conséquent une modification.
Paramètres influençant la plasticité
En général, les polymères ont la capacité de réorienter leurs chaînes à l’instar des autres matériaux (métaux, céramiques).
Leur comportement mécanique est donc très sensible à la température et à la vitesse de sollicitation, car ces paramètres influent sur la mobilité des chaînes. Ce sont ces deux paramètres principaux qui agissent par voie de conséquence sur la plasticité.
D’autre part, la structure des molécules joue également un rôle important sur les propriétés mécaniques. Par exemple, la longueur des chaînes et leurs enchevêtrements vont influer sur la mobilité moléculaire et donc sur la plasticité. Les conditions externes peuvent avoir aussi une influence sur le comportement mécanique.
La température
La température est un paramètre essentiel sur le comportement mécanique. C’est ce paramètre qui est pris en compte pour le tracé des cartes de résistance mécanique. Une telle dépendance s’explique par le fait que la mobilité moléculaire augmente énormément avec la température. En revanche, la limite élastique diminue progressivement quand on augmente la température.
Ainsi, le comportement mécanique des polymères à basse température (T< 0,8 Tg) est un comportement rigide et fragile. En revanche, le comportement des polymères est souple et ductile à haute température (T> Tg). Cela se traduit par l’absence de striction, d’instabilité plastique et une déformation homogène sous contrainte faible.
Dans la zone de transition ductile/fragile (T~0,8 Tg), l’amorce de la plasticité est une cohabitation des deux modes. Pour le polycarbonate, cela se manifeste par une instabilité plastique sans striction.
La vitesse de sollicitation
En ce qui concerne l’influence de la vitesse de sollicitation, l’évolution de comportement mécanique des polymères en vitesse de sollicitation est presque équivalente à l’évolution de comportement mécanique des polymères en température. Plus la vitesse de sollicitation est élevée, et plus les polymères sont rigides et fragiles. En revanche, ils sont souples et ductiles à vitesse de sollicitation lente.
La vitesse de sollicitation agit principalement sur la vitesse de déformation du polymère. Par conséquent au niveau des craquelures, plus la vitesse sera lente et plus les chaînes auront le temps de se réorienter au sein des craquelures pour créer des microcavités dans tout l’échantillon.
La pression hydrostatique
Entre les unités constitutives d’un polymère, il existe un volume, appelé volume libre, sur lequel l’exercice d’une pression modifie considérablement les propriétés mécaniques, en l’occurrence la contrainte critique.
La structure moléculaire
La nature et la structure d’un matériau polymère influent sur la plasticité. Par exemple, la longueur et la mobilité des chaînes, mais surtout les degrés d’enchevêtrement, de cristallinité et de réticulation agissent sur la faculté plus ou moins grande de pouvoir se déformer.
Également l’orientation moléculaire et la nature des renforts sont des paramètres influents. On peut prendre l’exemple d’un polymère ayant des groupements latéraux encombrants comme des groupes phényles et qui par conséquent aurait une mobilité plus faible qu’un polymère peu encombré. Ainsi, les cinétiques des phénomènes d’endommagement seraient différentes.
Les conditions de mise en œuvre interviennent également car la présence de défauts, de contraintes internes et l’état de surface créent une certaine plasticité supplémentaire.
L’environnement extérieur
Un autre paramètre pouvant influencer la plasticité des polymères est l’environnement dans lequel il est utilisé. En effet, la teneur en eau du milieu ainsi que la présence d’agents chimiques peuvent agir sur la résistance et la rigidité du matériau. Par exemple, la résistance et la rigidité diminue lorsque la teneur en eau augmente pour les polyamides.
De même, si le polymère est en contact avec un gaz ou s’il est irradié par des rayons α, rayons β ou rayons γ (radioactivité) ; cela aura des conséquences sur l’endommagement du polymère et le conduira à la ruine.
Loi d’Eyring
Tous les paramètres précédemment cités influent sur la plasticité des polymères. Ces paramètres entrent également en jeu dans la loi d’Eyring qui permet, comme les critères de plasticité, de décrire le comportement des polymères amorphes en déformation.
Cette approche de Ree-Eyring date de 1958 et a été développée par Duckett et Bauwens par la suite. Il s’agit essentiellement d’une loi phénoménologique traitant la déformation plastique comme un processus d’écoulement visqueux, activé à la fois par la température et le travail des contraintes. Ainsi, les caractéristiques moléculaires de la chaîne polymère ne sont pas seules responsables du processus de déformation.
Grâce à ce modèle, on est en mesure de calculer le seuil de plasticité en le reliant au temps de relaxation d’une portion de chaîne moléculaire ainsi qu’à la barrière d’énergie intermoléculaire.
On montre alors que :
- (4.1)

La pression hydrostatique influence le temps de relaxation des chaînes, il convient donc d’en tenir compte dans le calcul. On montre ainsi que la vitesse de déformation devient :
- (4.2)

D’après Bauwens-Crowet, on obtient pour le polycarbonate une série de droites parallèles pour diverses températures.
L'évolution linéaire de σy/T en fonction de log() confirme la loi de Eyring. Cependant, des mesures similaires sur le PMMA (ou le PVC) montrent l’obtention de courbes et non de droites.
Cette différence peut s’expliquer par l’existence de transitions secondaires omis dans les hypothèses de la théorie de Eyring. Cela revient à ne pas considérer le travail de la contrainte appliquée sur les groupements latéraux, ce qui est possible avec le PC qui possède des groupements latéraux relativement petits.
Notes et références
Annexes
Articles connexes
Bibliographie
- C. G’Sell, J-M. Haudin, Introduction à la mécanique des polymères, p. 321-337 et 395-402, (1995).
- C. Oudet, Polymères Structure et Propriétés, Introduction, Masson, p. 123-142, (1994).
- (en) E. Miller, Introduction to Plastics and Composites - Mechanical Properties and Engineering Applications, Marcel Dekker, p. 192-214, (1996) (ISBN 0-8247-9663-2).
- M. Fontanille, Y. Gnanou, Chimie et physico-chimie des polymères, Dunod, p. 426-427, (2002).
- G. Ehrenstein, F. Montagne, Matériaux polymères - structure propriétés et applications, Hermes Science Publishing, p. 133-147, (2000).
- H-H. Kausch, N. Heymans, C.J. Plummer, P. Decroly, Matériaux polymères : propriétés mécaniques et physiques, Traité des matériaux, tome 14, Presses Polytechniques et Universitaires Romandes, p. 275-388, (2001).
- (en) H-H. Kausch, Intrinsic Molecular Mobility and Toughness of Polymers, Springer Verlag, p. 1-35, (2005).
- (en) I.M. Ward, J. Sweeney, An introduction of the mechanical properties of solid polymers, second edition, John Wiley & Sons, p. 238-239 et 281-295, (1984).
- C. Oudet, Polymères, Structure et propriétés - Introduction, p. 132-142, (1994).
- P. Combette, I. Ernoutt, Physique des polymères, tome II, Propriétés mécaniques, Hermann, Ch XI, p. 337-384, (2005).