NF-κB
NF-κB pour nuclear factor-kappa B est une protéine de la superfamille des facteurs de transcription impliquée dans la réponse immunitaire et la réponse au stress cellulaire. Cette dernière est associée aux facteurs anti-apoptotiques. En effet son activation par la libération de sa protéine inhibitrice (IKB) déclenche la transcription de gènes anti-apoptotiques dans le noyau. Elle effectue donc un rétrocontrôle négatif de l'apoptose. C'est un sujet de recherche actuellement très étudié dans la mesure où plusieurs centaines de modulateurs de NF-κB sont connus et plus d'un millier de gènes cibles de ce facteur de transcription ont été identifiés.
Structure
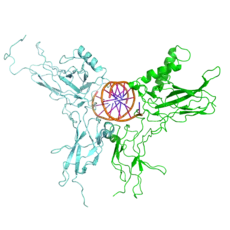
NF-κB est un homo- ou hétérodimère formé à partir de cinq sous-unités[1] : p50NF-κB1, p52NF-κB2, p65RelA, RelB et c-Rel. L’hétérodimère p50:p65 constitue la forme classique, la plus étudiée, de NF-κB.
Toutes les sous-unités sont caractérisées par un domaine N-terminal conservé d’environ 300 acides aminés[2], le Rel Homology Domain (RHD), contenant un domaine de liaison à l’ADN, un domaine de dimérisation, un signal de localisation nucléaire et un domaine d'interaction avec la protéine inhibitrice IκB. Les sous-unités RelA, RelB et c-Rel contiennent également un domaine de transactivation, responsable des activités de régulation transcriptionnelle de NF-κB.
Mécanisme d'action
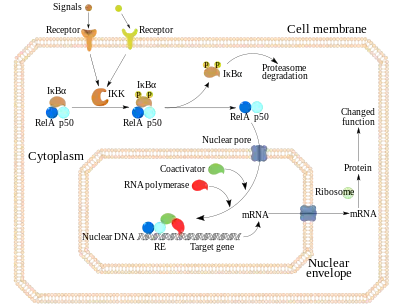
NF-κB est en partie régulée dans le cytoplasme de la cellule par un complexe protéique (IKK) composé des protéines, IKKα, IKKβ et de la protéine NEMO (IKKg). Elle joue un rôle dans la répression ou l'activation des gènes, et est retenue dans le cytoplasme par la protéine inhibitrice IκB-α. La dégradation de IκB-α par phosphorylation et ubiquitination permet la translocation de NF-κB jusqu'au noyau et l'activation de la transcription des gènes cibles.
Fonction
NF-κB est le pivot des cellules phagocytaires. Il permet de les activer. Il est activé grâce au complexe membranaire CD14-Toll-like receptor au contact d'un PAMP ; par exemple, Toll-like receptor 4 reconnait les LPS bactériens. Le complexe membranaire va en fait permettre la dégradation de IκB qui retient NF-κB dans sa forme inactive. Une fois libéré, il se dirige vers le noyau et permet la transcription de nouveaux gènes.
IκB peut être défini comme étant un séquestreur cytosolique. La séquestration cytosolique de NF-κB par IκB laisse le facteur NF-κB inactif (inhibition). En intra-cytosolique, NF-κB est lié au facteur IκB mais des stimuli externe peuvent permettre la dissociation du complexe NF-κB/IκB. Une autre kinase, IKK, pourra ainsi dans certains cas phosphoryler IκB ; NF-κB se retrouve donc libre et peut ainsi rentrer dans le noyau pour atteindre le génome (translocation nucléaire de NF-κB). De son côté, IκB est voué à la dégradation dans le protéasome 26S à la suite de l'accrochage d'une chaine d'ubiquitine.
Divers stimuli peuvent phosphoryler IκB et ainsi dissocier le complexe NF-κB/IκB. Ce qui, par conséquent, pourra activer NF-κB. Ces stimuli peuvent être physiologiques ou non : ce sont des carcinogènes, des facteurs pro-apoptotiques, des réactifs oxygénés, des cytokines, des facteurs de stress cellulaires (variés), des lipopolysaccharides bactériens, etc.
L'activation du facteur NF-κB peut se faire de deux façons : canonique et non-canonique.
Activation canonique
C'est la voie la plus simple. Un ligand extracellulaire se fixe à un type de récepteur membranaire (par exemple, des récepteurs TLR activés par divers types de ligands), ce qui entraîne un recrutement et une activation du complexe IKK. Pour rappel, IKK comprend IKKa, IKKb et NEMO.
Le complexe IKK activé phosphoryle alors IKB (IKB étant le séquestreur cytosolique de NF-κB) qui sera ensuite envoyé vers le protéasome 26S pour subir une dégradation. NF-κB peut ainsi être transloqué dans le noyau. La voie canonique active le plus souvent les dimères NF-κB comprenant Rel-A, c-Rel, Rel-B et p50.
Activation non canonique
Elle inclut l'activation des complexes p100/RelB et s'observe souvent durant le développement de certains organes lymphoïdes (génération et formation des lymphocytes B et T). Actuellement, on ne connaît que très peu de stimuli permettant de l'activer (lymphotoxine B, B cell activating factor, etc).
La caractéristique de la voie non canonique est l'utilisation d'un complexe IKK ayant deux sous-unités IKKa sans NEMO.
Le récepteur activé induira l'activation de la protéine NIK (NF-κB inducing kinase), qui phosphoryle et active le complexe IKKa, qui à son tour phosphoryle p100. Il s'ensuit une libération d'un hétérodimère actif p52/Rel-B. Contrairement au facteur p100, p105 subit un clivage pour donner p50.
Régulation du facteur IκB et de l'action de NF-κB
Les IKK sont des IκB-kinases. La régulation du facteur IκB se fait essentiellement par des phosphorylations sur les résidus Ser32 et Ser36, grâce aux IKK. IKK est fait de l'association d'un hétérodimère NEMO (constitué de IKKa et IKKb) avec une sous-unité IKKy.
Les IKK sont activés par de nombreux couples ligand-récepteurs, variés également. Parmi eux, on peut citer :
- Le TNF-α avec le TNF-Receptor : l'activation du TNF-Receptor pourrait permettre l'activation de NF-κB qui dans ce cas stimule le facteur mTOR et inhibe la production de réactifs oxygénés cellulaires, ce qui serait à la base d'un blocage des processus d'autophagie, notamment dans les cellules cancéreuses[3].
- L'IL-1β avec les lipopolysaccharides bactériens ; en effet, l'IL-1β peut se fixer sur son IL-1βR du macrophage (récepteur de cette cytokine inflammatoire) qui stimulera IKK. Cela engendre l'activation de NF-κB (dimère p50/p65) qui stimulera la transcription du gène de la NOS inductible. Celle-ci sera ensuite sécrétée par le macrophage afin d'aller bloquer le métabolisme mitochondrial des bactéries, ce qui engendre la mort de la bactérie. C'est un processus très utilisé lors de l'inflammation. L'interaction du lipopolysaccharide bactérien ou du lipopolysaccharide binding protein (LBP) avec le récepteur CD14 (en surface du macrophage) peut enclencher des voies de signalisation très similaires.
- Le TLR4 (CD 284), récepteur pouvant être activé par les lipopolysaccharides de bactéries Gram -, ce qui activera la voie MyD88 en intracytosolique afin de stimuler une activation indirecte (pouvant être Akt-dépendante) du facteur NF-κB.
- L'hypoxie est capable, selon les cas, de stimuler la production de réactifs oxygénés qui peuvent stimuler l'activation de NF-κB et de facteurs HIF-1α et ensemble, stimuleront l'autophagie.
- La kinase Akt concourt indirectement à l'activation de NF-κB : Akt peut en effet activer le complexe IKK par phosphorylation, qui lui-même pourra phosphoryler IKB, ce qui activera le facteur NF-κB.
- Divers antigènes activant les B-cell Receptor (BCR) ou T-cell Receptor (TCR) : par exemple, une infection bactérienne peut stimuler la production d'Interleukine 2 en activant la voie incluant le complexe NFAT/AP-1/NF-κB. NFAT est activé par le Ca2+, AP-1 est activé par des MAP kinases et la protéine kinase C (PKC) activera NF-κB. L'association des trois facteurs pourra se fixer sur le promoteur du gène de l'interleukine 2 et ainsi stimuler sa transcription. La sécrétion de l'IL-2 par les cellules T est une caractéristique majeure de la réponse immune adaptative.
- Le signal costimulateur des lymphocytes T permet également d'accroître le taux de NF-κB libre. En effet l'activation du CD28 (de la cellule T) par les CD80 et CD86 (aussi connus sous le nom de B7.1 ET B7.2) de la CPA permet l'augmentation de la production des facteurs AP-1 et NF-κB, triplant ainsi la transcription de l'ARNm de l'IL-2[4].
Physiopathologie
- Son activation exagérée (lors de la présence de bactéries dans le sang par exemple) peut provoquer un choc septique.
- NF-κB est connu pour être un des multiples régulateurs de plusieurs gènes codant des protéines de l'inflammation ; ce facteur dispose ainsi d'une fenêtre de temps d'activation (temps passé dans le noyau) beaucoup plus long que la normale dans diverses pathologies inflammatoires comme : arthrite, maladies inflammatoires de l'intestin, asthme, athérosclérose, etc[5].
- Il a été montré dans de très nombreuses publications l'implication du facteur NF-κB dans la cancérogenèse et les processus de tumorisation de certaines cellules[6] - [7].
Notes et références
- Pereira et Oakley 2008
- Perkins et Gilmore 2006
- Autophagy and NF-κB signalling pathways in cancer cells
- Immunobiologie, Éditions De Boeck, 2009 - page 345.
- ^ Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, Cheshire N, Paleolog E, Feldmann M (April 2004). "Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis". Proc. Natl. Acad. Sci. U.S.A. 101 (15): 5634–9. doi:10.1073/pnas.0401060101. PMC 397455.
- Hoesel, Bastian; Schmid, Johannes A.. The complexity of NF-κB signaling in inflammation and cancer. Molecular Cancer, 2013 12:86
- (en) N. Perkins et T. Gilmore, « Good cop, bad cop: the different faces of NF-kappaB », Cell Death Differ., vol. 13, no 5, , p. 759–772 (PMID 16410803, DOI 10.1038/sj.cdd.4401838)
Voir aussi
Bibliographie
- (en) Silvia Pereira et Fiona Oakley, « Nuclear factor-kappaB1: regulation and function », Int. J. Biochem. Cell Biol., vol. 40, no 8, , p. 1425–1430 (PMID 17693123, DOI 10.1016/j.biocel.2007.05.004)