Maladie de Griscelli
La maladie ou le syndrome de Griscelli (SG) a été décrit pour la première fois par Griscelli et Pruniéras en 1978[1]. Le SG est une maladie héréditaire rare caractérisée par un albinisme partiel associé à d’autres pathologies dans les cas les plus graves. C’est une maladie génétique à transmission autosomique récessive due à des mutations des gènes codant le complexe transporteur des mélanosomes.
Prévalence
La plupart des cas sont originaires du pourtour méditerranéen dont essentiellement la Turquie, toutefois quelques cas originaires d’Inde ont été décrits en 2004[2]. Soixante cas étaient décrits dans le monde entier au [3].
Cette maladie touche aussi bien les hommes que les femmes.
Description clinique
Le syndrome de Griscelli est classé en trois types[1] - [4] tous caractérisés par la présence de cheveux aux reflets gris, argentés (on parle parfois de reflets dorés ou poussiéreux), mais également par la présence d’une hypopigmentation au niveau cutané caractérisant un albinisme[5]. De plus, on observe dans le syndrome de type 1 des atteintes neurologiques : convulsions, paralysie, spasticité. Le patient souffrira d’un retard psychomoteur important. Le type 2 se caractérise par une immunodéficience (pouvant également déboucher sur des problèmes neurologiques). Cette immunodéficience est à l’origine d’infections diverses au niveau cutané, ORL, respiratoire. Elle se caractérise par une leucopénie et une thrombopénie. En outre, il peut se produire une activation macrophagique (syndrome hémophagocytaire) c'est-à-dire qu’il y aura phagocytose des éléments sanguins et libération de cytokines post inflammatoires. Des lymphocytes T pourront également s’infiltrer dans le cerveau. Une hépatosplénomégalie ainsi que des problèmes oculaires peuvent également apparaitre.
Diagnostic

Les méthodes de diagnostic sont basées sur le principe du diagnostic différentiel : le but est d’éliminer peu à peu les autres maladies ayant des symptômes proches de ceux de la maladie de Griscelli. L’étude microscopique des cheveux montre une répartition irrégulière (en motte) des pigments. La microscopie électronique à transmission révèle que les mélanocytes sont surchargés de mélanosomes : organites fabriquant la mélanine, au contraire les kératinocytes contiennent très peu de mélanosomes. Les frottis sanguins révèlent l’absence de granules cytoplasmiques dans les leucocytes. Les électroencéphalogrammes peuvent mettre en évidence les problèmes neurologiques. Des biopsies de foie et de moelle osseuse sont également utilisées pour le diagnostic. Par ailleurs, pour les types 1 et 2 il existe un diagnostic anténatal c’est le prélèvement de villosités choriales qui sera suivi d’un séquençage des gènes
Physiopathologie
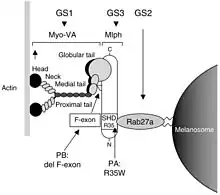
Les trois syndromes de Griscelli se rejoignent dans l'hypopigmentation de la peau. La pigmentation de la peau est effectuée par des cellules spécialisées productrices de mélanine, les mélanocytes. Les mélanocytes se trouvent à la base de l’épiderme, et proviennent de l'ectoderme embryonnaire. Le corps cellulaire est arrondi et porte de nombreux prolongements ramifiés qui s'insinuent entre les kératinocytes de la couche supérieure. Les prolongements, appelés dendrites mélanocytaires, lui permettent de communiquer avec les kératinocytes qui synthétisent la kératine. Les mélanocytes fabriquent des petits sacs, les mélanosomes, contenant de la mélanine qui est le pigment foncé. Chaque mélanocyte prend en charge la protection de 36 kératinocytes formant ainsi une unité épidermique de mélanisation. Il leur transfère ses mélanosomes le long des filaments d'actines grâce à un complexe transporteur composé de trois unités codés par les gènes MYO5A, RAB27A et le gène MLPH[2] - [6].
Syndrome de Griscelli de type 1 (SG1)
La cause du SG1 résulte d'une mutation du gène de la myosine 5a (MYO5A), situé sur le chromosome 15q21[7]. Le SG1 correspond probablement au syndrome d'Elejalde (en). Le gène MYO5A code la myosine 5a, une protéine motrice se liant à l'actine et jouant un rôle dans le transport intracellulaire des mélanosomes dans les dendrites des mélanocytes. Ainsi une mutation de ce gène empêche le transport des mélanosomes. Le retard psychomoteur précoce et sévère est peu expliqué cependant on pense qu'il serait dû à problème de transport du réticulum endoplasmique dans les dendrites des neurones.
Syndrome de Griscelli de type 2 (SG2)
Certains ganglions et organes (dont le cerveau) sont infiltrés par des lymphocytes T et des macrophages qui une fois activés phagocytent les cellules sanguines c'est le syndrome hémophagocytaire. Le SG2 résulte d'une mutation du gène RAB27A (localisé dans la même région que le gène MYO5A)[8]. Il code d'une part des effecteurs clés du transport vésiculaire intracellulaire. Donc en cas de mutation le transport vésiculaire des mélanosomes s'avère modifié. D'autre part, la protéine Rab27a régule aussi la sécrétion des granules cytotoxiques d'où le déclenchement du syndrome hémophagocytaire[9].
Syndrome de Griscelli de type 3 (SG3)
Le SG3 est dû à des mutations de MLPH, un gène codant la mélanophiline, qui forme un complexe protéique avec Rab27a et la myosine 5a et participe au transport des mélanosomes dans les mélanocytes.
Prise en charge thérapeutique et pronostic
Pour le SG1, le traitement reste symptomatique et limité avec la prise de corticostéroïdes, antiépileptiques, et antipyrétiques. Toutefois, cette prise en charge ne parvient généralement pas à empêcher le décès précoce provoqué par l'atteinte neurologique sévère.
Pour le SG2, Le syndrome hémophagocytaire est souvent fatal et le seul traitement curatif est la greffe de moelle osseuse[10].
Pour le SG3, à ce jour aucun traitement n’est proposé.
Le pronostic de survie des patients atteint du syndrome de Griscelli de type 1 et 2 est relativement faible, d'habitude, la maladie est fatale dans les 1 à 4 ans sans traitement. Mais la maladie est plus viable pour le patient atteint d'un SG de type 3, car il n'a que des problèmes de peau superficiels[11].
Notes et références
- (en) Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, Prunieras M. « A syndrome associating partial albinism and immunodeficiency » Am J Med. 1978;65:691-702.
- (en) Martino Ruggieri, Ignacio Pascual Castroviejo, Concezio Di Rocco, Neurocutaneous Disorders: Phakomatoses & Hamartoneoplastic Syndromes, Spinger Wien NewYork (ISBN 978-3211213964). p. 418-426
- (en) Fischer A, Virelizier JL, Arenzana SF et al. « Treatment of four patients with erythrophagocytosis by a combination of epipodophyllotoxin, steroids, intracranial methotrexate and cranial irradiation » Pediatrics 1985;76:263-8.
- page spécifique sur le site Orphanet.
- (en) Rajadhyax M, Neti G, Crow Y, Tyagi A. « Neurological presentation of Griscelli syndrome: Obstructive hydrocephalus without hematological abnormalities or organomegaly » Brain Dev. 2007;29:247-50.
- (en) « Lippincott Home », sur jisppd.com (consulté le ).
- (en) Klein C, Philippe N, Le Deist F, Fraitag S, Prost C, Durandy A, Fischer A, Griscelli C. « Partial albinism with immunodeficiency » J Pediatr. 1994; 125:886-95.
- (en) Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, Wulffraat N, Bianchi D, Fischer A, Le Deist F, de Saint BG. « Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome » Nat Genet. 2000;25:173-6.
- Syndrome hémophagocytaire dans une autre revue de pédiatrie.
- (en) Hurvitz H, Gillis R, Klaus R et al. « Kindred with Griscelli disease: spectrum of neurological involvement » Eur J Pediatr. 1993;152:402-5.
- http://www.jpad.org.pk/April%20June%202007/11.%20Case%20report%20Griscelli%E2%80%99s%20syndrome.pdf.
- (en) M Seiberg, « Keratinocyte-melanocyte interactions during melanosome transfer - PubMed », Pigment cell research, vol. 14, no 4, , p. 236–242 (ISSN 0893-5785, PMID 11549105, DOI 10.1034/j.1600-0749.2001.140402.x, lire en ligne, consulté le )
- http://despedara.org/cours_des/syndrome_griscelli_30_11_2007.pdf