Méthode de Joback
La méthode de Joback[1] (aussi connue sous le nom de méthode de Joback-Reid) prédit onze propriétés thermodynamiques importantes et couramment utilisées pour des composés purs à partir de leur seule structure moléculaire.
Fondements
Méthode de contribution de groupes
La méthode de Joback est une méthode de contribution de groupes (en), c'est-à-dire qu'elle utilise les informations structurelles de base d'une molécule chimique, par exemple une liste de groupes fonctionnels simples, associe des paramètres à ces groupes fonctionnels et calcule les propriétés physico-chimiques du composé en fonction de la somme des paramètres de groupes.
Cette méthode suppose qu'il n'y a pas d'interactions entre les groupes, et ne fait donc que sommer les contributions de chaque groupe sans ajouter de terme pour les interactions entre les groupes. D'autres méthodes de contribution de groupes, en particulier des méthodes comme UNIFAC, qui estime les propriétés du mélange comme les coefficients d'activité, utilisent à la fois de simples paramètres additifs de groupes et des paramètres d'interaction entre groupes. L'utilisation de paramètres de groupes simples présente néanmoins l'avantage de ne nécessiter qu'un petit nombre de paramètres. : le nombre de paramètres d'interaction de groupe à prendre en compte devient en effet très élevé pour un nombre croissant de groupes (un seul pour deux groupes, trois pour trois groupes, six pour quatre groupes, 45 pour dix groupes et deux fois plus si les interactions ne sont pas symétriques).
Neuf des propriétés calculables par la méthode de Joback sont des constantes indépendantes de la température, principalement estimées par une simple somme des contributions des groupes et d'un terme-source. Deux des propriétés estimées dépendent de la température : la capacité thermique du gaz parfait et la viscosité dynamique des liquides. Le polynôme utilisé pour calculer la capacité thermique utilise quatre paramètres et l'équation de viscosité seulement deux. Dans les deux cas, les paramètres de l'équation sont obtenus en sommant les contributions des groupes.
Histoire
La méthode de Joback est une extension de la méthode de Lydersen (en)[2] et utilise des groupes, des formules et des paramètres très similaires pour les trois propriétés déjà couvertes par celle-ci (température, pression et volume critiques).
Joback élargit la gamme des propriétés prises en charge, créé de nouveaux paramètres et modifié légèrement les formules de son prédécesseur.
Avantages et inconvénients du modèle
Avantages
La popularité et le succès de la méthode de Joback proviennent principalement de la liste de groupe unique pour toutes les propriétés, qui permet d'obtenir les onze propriétés prises en charge à partir d'une seule analyse de la structure moléculaire.
La méthode de Joback utilise en outre un schéma de groupe très simple et facile à appliquer, ce qui rend la méthode utilisable pour les personnes n'ayant que des connaissances chimiques de base.
Inconvénients
Par rapport aux développements ultérieurs des méthodes d'estimation[3] - [4], la qualité de la méthode de Joback est limitée. Les auteurs originaux le déclaraient déjà dans le résumé de l'article original: « Nous ne prétendons pas à un haut degré d'exactitude, mais la précision des méthodes que nous proposons est souvent égale ou supérieure aux techniques communément utilisées aujourd'hui. »
La liste des groupes offre une couverture limitée pour de nombreuses molécules pourtant courantes. En particulier, les composés aromatiques ne sont pas différenciés des composés normaux contenant un cycle, ce qui est problématique car les propriétés des aromatiques et aliphatiques sont en général assez différentes.
La base de données utilisée par Joback et Reid pour obtenir les paramètres de groupe était relativement restreinte et ne couvrait qu'un nombre limité de molécules. La meilleure couverture a été obtenue pour les points d'ébullition (438 composés) et la moins bonne pour les enthalpies de fusion (155 composés). Les méthodes actuelles peuvent s'appuyer sur des banques de données, comme la banque de données de Dortmund (en)ou la base de données DIPPR, qui offrent une couverture beaucoup plus large.
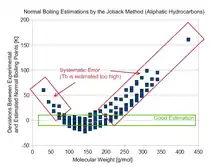
La formule utilisée pour la prédiction du point d'ébullition à pression atmosphérique présente un autre problème : Joback suppose que les contributions des groupes sommés sont constantes dans des séries homologues comme les alcanes, or ce postulat ne décrit pas le comportement réel des points d'ébullition[5], qui devrait être modélisé avec des contributions de plus en plus faibles pour chaque groupe à mesure que le nombre de groupes augmente. La formule utilisée dans la méthode de Joback conduit ainsi à des écarts importants pour les molécules relativement grandes ou petites, avec une estimation plutôt bonne pour les composés de taille moyenne seulement.
Formules
Dans les formules suivantes, Gi désigne une contribution de groupe. Les Gi sont comptés pour chaque groupe disponible. Si un groupe est présent plusieurs fois, chaque occurrence est comptée séparément.
Point d'ébullition à pression atmosphérique
![{\displaystyle T_{\text{eb}}[{\text{K}}]=198.2+\sum T_{{\text{eb}},i}.}](https://img.franco.wiki/i/43f577a9a663d17ab02395b3e01cb1887ea9b4b2.svg)
Point de fusion
![{\displaystyle T_{\text{fus}}[{\text{K}}]=122.5+\sum T_{{\text{fus}},i}.}](https://img.franco.wiki/i/86265287dea5e64080b312d043aa4b093169ea6a.svg)
Température critique
![{\displaystyle T_{\text{c}}[{\text{K}}]=T_{\text{eb}}\left[0.584+0.965\sum T_{{\text{c}},i}-\left(\sum T_{{\text{c}},i}\right)^{2}\right]^{-1}.}](https://img.franco.wiki/i/75b69444256a1096880458807784b2f4b0f2802d.svg)
Cette équation de température critique nécessite un point d'ébullition à pression atmosphérique Teb . Si une valeur expérimentale est disponible, il est recommandé d'utiliser ce point d'ébullition. Il est également possible de se servir du point d'ébullition estimé par la méthode de Joback, ce qui conduit toutefois à une erreur plus élevée.
Pression critique
![{\displaystyle P_{\text{c}}[{\text{bar}}]=\left[0.113+0.0032\,N_{\text{a}}-\sum P_{{\text{c}},i}\right]^{-2},}](https://img.franco.wiki/i/85925f3f1489f0f5550018673c083de4449d0f24.svg)
où Na est le nombre d'atomes dans la molécule (y compris les hydrogènes).
Volume critique
![{\displaystyle V_{\text{c}}[{\text{cm}}^{3}/{\text{mol}}]=17.5+\sum V_{{\text{c}},i}.}](https://img.franco.wiki/i/f6cd4837150b1524a1968d7508a93289fc6d3133.svg)
Enthalpie de formation (gaz parfait, 298 K)
![{\displaystyle H_{\text{formation}}[{\text{kJ}}/{\text{mol}}]=68.29+\sum H_{{\text{form}},i}.}](https://img.franco.wiki/i/8ff4df9e7e76e9ed5d5635acf868cbb74e700b89.svg)
Enthalpie libre de formation (gaz parfait, 298 K)
![{\displaystyle G_{\text{formation}}[{\text{kJ}}/{\text{mol}}]=53.88+\sum G_{{\text{form}},i}.}](https://img.franco.wiki/i/88f1922d8cfd2d9c88afdebe36584f9557636999.svg)
Capacité calorifique (gaz parfait)
![{\displaystyle C_{P}[{\text{J}}/({\text{mol}}\cdot {\text{K}})]=\sum a_{i}-37.93+\left[\sum b_{i}+0.210\right]T+\left[\sum c_{i}-3.91\cdot 10^{-4}\right]T^{2}+\left[\sum d_{i}+2.06\cdot 10^{-7}\right]T^{3}.}](https://img.franco.wiki/i/5ae2f45d0ad4e1358033466231bedc7e4c6971b7.svg)
La méthode de Joback utilise une modélisation polynomiale à quatre paramètres pour décrire l'évolution de la capacité calorifique du gaz parfait en fonction de la température. Ces paramètres sont valables à partir de 273 K et jusqu'à environ 1 000 K.
Enthalpie de vaporisation au point d'ébullition atmosphérique
![{\displaystyle \Delta H_{\text{vap}}[{\text{kJ}}/{\text{mol}}]=15.30+\sum H_{{\text{vap}},i}.}](https://img.franco.wiki/i/25c2ac235c1bd0109b3670a794306a1053466ac7.svg)
Enthalpie de fusion
![{\displaystyle \Delta H_{\text{fus}}[{\text{kJ}}/{\text{mol}}]=-0.88+\sum H_{{\text{fus}},i}.}](https://img.franco.wiki/i/3885a8777dde47f4a1e7aa4e957e302380424d5e.svg)
Viscosité dynamique à l'état liquide
![{\displaystyle \eta _{\text{L}}[{\text{Pa}}\cdot {\text{s}}]=M_{\text{w}}exp{\left[\left(\sum \eta _{a}-597.82\right)/T+\sum \eta _{b}-11.202\right]},}](https://img.franco.wiki/i/dc922223e7bf93e11d34cbd9caa9d50ba7f0b846.svg)
où Mw est la masse moléculaire.
La méthode utilise une équation à deux paramètres pour décrire l'évolution de la viscosité dynamique en fonction de la température. Les auteurs précisent que les paramètres sont valables de la température de fusion jusqu'à 70 % de la température critique (Tr < 0,7).
Contributions de groupes
| Groupe | Tc | Pc | Vc | Teb | Tfus | Hform | Gform | a | b | c | d | Hfusion | Hvap | ηa | ηb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Données au point critique | Températures de changement d'état | Propriétés thermochimiques | Capacités calorifiques du gaz parfait | Enthalpies de changement d'état | Viscosité dynamique | ||||||||||
| Groupes non cycliques | |||||||||||||||
| −CH3 | 0.0141 | −0.0012 | 65 | 23.58 | −5.10 | −76.45 | −43.96 | 1.95E+1 | −8.08E−3 | 1.53E−4 | −9.67E−8 | 0.908 | 2.373 | 548.29 | −1.719 |
| −CH2− | 0.0189 | 0.0000 | 56 | 22.88 | 11.27 | −20.64 | 8.42 | −9.09E−1 | 9.50E−2 | −5.44E−5 | 1.19E−8 | 2.590 | 2.226 | 94.16 | −0.199 |
| >CH− | 0.0164 | 0.0020 | 41 | 21.74 | 12.64 | 29.89 | 58.36 | −2.30E+1 | 2.04E−1 | −2.65E−4 | 1.20E−7 | 0.749 | 1.691 | −322.15 | 1.187 |
| >C< | 0.0067 | 0.0043 | 27 | 18.25 | 46.43 | 82.23 | 116.02 | −6.62E+1 | 4.27E−1 | −6.41E−4 | 3.01E−7 | −1.460 | 0.636 | −573.56 | 2.307 |
| =CH2 | 0.0113 | −0.0028 | 56 | 18.18 | −4.32 | −9.630 | 3.77 | 2.36E+1 | −3.81E−2 | 1.72E−4 | −1.03E−7 | −0.473 | 1.724 | 495.01 | −1.539 |
| =CH− | 0.0129 | −0.0006 | 46 | 24.96 | 8.73 | 37.97 | 48.53 | −8.00 | 1.05E−1 | −9.63E−5 | 3.56E−8 | 2.691 | 2.205 | 82.28 | −0.242 |
| =C< | 0.0117 | 0.0011 | 38 | 24.14 | 11.14 | 83.99 | 92.36 | −2.81E+1 | 2.08E−1 | −3.06E−4 | 1.46E−7 | 3.063 | 2.138 | n. a. | n. a. |
| =C= | 0.0026 | 0.0028 | 36 | 26.15 | 17.78 | 142.14 | 136.70 | 2.74E+1 | −5.57E−2 | 1.01E−4 | −5.02E−8 | 4.720 | 2.661 | n. a. | n. a. |
| ≡CH | 0.0027 | −0.0008 | 46 | 9.20 | −11.18 | 79.30 | 77.71 | 2.45E+1 | −2.71E−2 | 1.11E−4 | −6.78E−8 | 2.322 | 1.155 | n. a. | n. a. |
| ≡C− | 0.0020 | 0.0016 | 37 | 27.38 | 64.32 | 115.51 | 109.82 | 7.87 | 2.01E−2 | −8.33E−6 | 1.39E-9 | 4.151 | 3.302 | n. a. | n. a. |
| Groupes cycliques | |||||||||||||||
| −CH2− | 0.0100 | 0.0025 | 48 | 27.15 | 7.75 | −26.80 | −3.68 | −6.03 | 8.54E−2 | −8.00E−6 | −1.80E−8 | 0.490 | 2.398 | 307.53 | −0.798 |
| >CH− | 0.0122 | 0.0004 | 38 | 21.78 | 19.88 | 8.67 | 40.99 | −2.05E+1 | 1.62E−1 | −1.60E−4 | 6.24E−8 | 3.243 | 1.942 | −394.29 | 1.251 |
| >C< | 0.0042 | 0.0061 | 27 | 21.32 | 60.15 | 79.72 | 87.88 | −9.09E+1 | 5.57E−1 | −9.00E−4 | 4.69E−7 | −1.373 | 0.644 | n. a. | n. a. |
| =CH− | 0.0082 | 0.0011 | 41 | 26.73 | 8.13 | 2.09 | 11.30 | −2.14 | 5.74E−2 | −1.64E−6 | −1.59E−8 | 1.101 | 2.544 | 259.65 | −0.702 |
| =C< | 0.0143 | 0.0008 | 32 | 31.01 | 37.02 | 46.43 | 54.05 | −8.25 | 1.01E−1 | −1.42E−4 | 6.78E−8 | 2.394 | 3.059 | -245.74 | 0.912 |
| Groupes halogènes | |||||||||||||||
| −F | 0.0111 | −0.0057 | 27 | −0.03 | −15.78 | −251.92 | −247.19 | 2.65E+1 | −9.13E−2 | 1.91E−4 | −1.03E−7 | 1.398 | −0.670 | n. a. | n. a. |
| −Cl | 0.0105 | −0.0049 | 58 | 38.13 | 13.55 | −71.55 | −64.31 | 3.33E+1 | −9.63E−2 | 1.87E−4 | −9.96E−8 | 2.515 | 4.532 | 625.45 | −1.814 |
| −Br | 0.0133 | 0.0057 | 71 | 66.86 | 43.43 | −29.48 | −38.06 | 2.86E+1 | −6.49E−2 | 1.36E−4 | −7.45E−8 | 3.603 | 6.582 | 738.91 | −2.038 |
| −I | 0.0068 | −0.0034 | 97 | 93.84 | 41.69 | 21.06 | 5.74 | 3.21E+1 | −6.41E−2 | 1.26E−4 | −6.87E−8 | 2.724 | 9.520 | 809.55 | −2.224 |
| Groupes oxygène | |||||||||||||||
| −OH (alcohol) | 0.0741 | 0.0112 | 28 | 92.88 | 44.45 | −208.04 | −189.20 | 2.57E+1 | −6.91E−2 | 1.77E−4 | −9.88E−8 | 2.406 | 16.826 | 2173.72 | −5.057 |
| −OH (phenol) | 0.0240 | 0.0184 | −25 | 76.34 | 82.83 | −221.65 | −197.37 | −2.81 | 1.11E−1 | −1.16E−4 | 4.94E−8 | 4.490 | 12.499 | 3018.17 | −7.314 |
| −O− (non cyclique) | 0.0168 | 0.0015 | 18 | 22.42 | 22.23 | −132.22 | −105.00 | 2.55E+1 | −6.32E−2 | 1.11E−4 | −5.48E−8 | 1.188 | 2.410 | 122.09 | −0.386 |
| −O− (cyclique) | 0.0098 | 0.0048 | 13 | 31.22 | 23.05 | −138.16 | −98.22 | 1.22E+1 | −1.26E−2 | 6.03E−5 | −3.86E−8 | 5.879 | 4.682 | 440.24 | −0.953 |
| >C=O (non cyclique) | 0.0380 | 0.0031 | 62 | 76.75 | 61.20 | −133.22 | −120.50 | 6.45 | 6.70E−2 | −3.57E−5 | 2.86E−9 | 4.189 | 8.972 | 340.35 | −0.350 |
| >C=O (cyclique) | 0.0284 | 0.0028 | 55 | 94.97 | 75.97 | −164.50 | −126.27 | 3.04E+1 | −8.29E−2 | 2.36E−4 | −1.31E−7 | 0. | 6.645 | n. a. | n. a. |
| O=CH− (aldéhyde) | 0.0379 | 0.0030 | 82 | 72.24 | 36.90 | −162.03 | −143.48 | 3.09E+1 | −3.36E−2 | 1.60E−4 | −9.88E−8 | 3.197 | 9.093 | 740.92 | −1.713 |
| −COOH (acide) | 0.0791 | 0.0077 | 89 | 169.09 | 155.50 | −426.72 | −387.87 | 2.41E+1 | 4.27E−2 | 8.04E−5 | −6.87E−8 | 11.051 | 19.537 | 1317.23 | −2.578 |
| −COO− (ester) | 0.0481 | 0.0005 | 82 | 81.10 | 53.60 | −337.92 | −301.95 | 2.45E+1 | 4.02E−2 | 4.02E−5 | −4.52E−8 | 6.959 | 9.633 | 483.88 | −0.966 |
| =O (autre que ci-dessus) | 0.0143 | 0.0101 | 36 | −10.50 | 2.08 | −247.61 | −250.83 | 6.82 | 1.96E−2 | 1.27E−5 | −1.78E−8 | 3.624 | 5.909 | 675.24 | −1.340 |
| Groupes azotés | |||||||||||||||
| −NH2 | 0.0243 | 0.0109 | 38 | 73.23 | 66.89 | −22.02 | 14.07 | 2.69E+1 | −4.12E−2 | 1.64E−4 | −9.76E−8 | 3.515 | 10.788 | n. a. | n. a. |
| >NH (non cyclique) | 0.0295 | 0.0077 | 35 | 50.17 | 52.66 | 53.47 | 89.39 | −1.21 | 7.62E−2 | −4.86E−5 | 1.05E−8 | 5.099 | 6.436 | n. a. | n. a. |
| >NH (cyclique) | 0.0130 | 0.0114 | 29 | 52.82 | 101.51 | 31.65 | 75.61 | 1.18E+1 | −2.30E−2 | 1.07E−4 | −6.28E−8 | 7.490 | 6.930 | n. a. | n. a. |
| >N− (non cyclique) | 0.0169 | 0.0074 | 9 | 11.74 | 48.84 | 123.34 | 163.16 | −3.11E+1 | 2.27E−1 | −3.20E−4 | 1.46E−7 | 4.703 | 1.896 | n. a. | n. a. |
| −N= (non cyclique) | 0.0255 | -0.0099 | n. a. | 74.60 | n. a. | 23.61 | n. a. | n. a. | n. a. | n. a. | n. a. | n. a. | 3.335 | n. a. | n. a. |
| −N= (cyclique) | 0.0085 | 0.0076 | 34 | 57.55 | 68.40 | 55.52 | 79.93 | 8.83 | −3.84E-3 | 4.35E−5 | −2.60E−8 | 3.649 | 6.528 | n. a. | n. a. |
| =NH | n. a. | n. a. | n. a. | 83.08 | 68.91 | 93.70 | 119.66 | 5.69 | −4.12E−3 | 1.28E−4 | −8.88E−8 | n. a. | 12.169 | n. a. | n. a. |
| −CN | 0.0496 | −0.0101 | 91 | 125.66 | 59.89 | 88.43 | 89.22 | 3.65E+1 | −7.33E−2 | 1.84E−4 | −1.03E−7 | 2.414 | 12.851 | n. a. | n. a. |
| −NO2 | 0.0437 | 0.0064 | 91 | 152.54 | 127.24 | −66.57 | −16.83 | 2.59E+1 | −3.74E−3 | 1.29E−4 | −8.88E−8 | 9.679 | 16.738 | n. a. | n. a. |
| Groupes sulfurés | |||||||||||||||
| −SH | 0.0031 | 0.0084 | 63 | 63.56 | 20.09 | −17.33 | −22.99 | 3.53E+1 | −7.58E−2 | 1.85E−4 | −1.03E−7 | 2.360 | 6.884 | n. a. | n. a. |
| −S− (non cyclique) | 0.0119 | 0.0049 | 54 | 68.78 | 34.40 | 41.87 | 33.12 | 1.96E+1 | −5.61E−3 | 4.02E−5 | −2.76E−8 | 4.130 | 6.817 | n. a. | n. a. |
| −S− (cyclique) | 0.0019 | 0.0051 | 38 | 52.10 | 79.93 | 39.10 | 27.76 | 1.67E+1 | 4.81E−3 | 2.77E−5 | −2.11E−8 | 1.557 | 5.984 | n. a. | n. a. |
Exemple de calcul
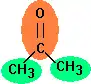
L'acétone (propanone) est la cétone la plus simple, divisée en trois groupes dans la méthode de Joback: deux groupes méthyle (-CH3 ) et un groupe cétone (C=O). Le groupe méthyle étant présent deux fois, ses contributions doivent être ajoutées deux fois.
| −CH3 | >C=O (non-ring) | ||||||
| Propriété | Nombre de groupes | Contribution de groupe | Nombre de groupes | Contribution de groupe | Valeur estimée | Unité | |
| Tc | 2 |
0.0141 |
1 |
0.0380 |
0.0662 |
500.5590 |
K |
| Pc | 2 |
−1.20E−03 |
1 |
3.10E−03 |
7.00E−04 |
48.0250 |
bar |
| Vc | 2 |
65.0000 |
1 |
62.0000 |
192.0000 |
209.5000 |
mL/mol |
| Tb | 2 |
23.5800 |
1 |
76.7500 |
123.9100 |
322.1100 |
K |
| Tm | 2 |
−5.1000 |
1 |
61.2000 |
51.0000 |
173.5000 |
K |
| Hformation | 2 |
−76.4500 |
1 |
−133.2200 |
−286.1200 |
−217.8300 |
kJ/mol |
| Gformation | 2 |
−43.9600 |
1 |
−120.5000 |
−208.4200 |
−154.5400 |
kJ/mol |
| Cp: a | 2 |
1.95E+01 |
1 |
6.45E+00 |
4.55E+01 |
||
| Cp: b | 2 |
−8.08E−03 |
1 |
6.70E−02 |
5.08E−02 |
||
| Cp: c | 2 |
1.53E−04 |
1 |
−3.57E−05 |
2.70E−04 |
||
| Cp: d | 2 |
−9.67E−08 |
1 |
2.86E−09 |
−1.91E−07 |
||
| Cp | à T = 300 K |
75.3264 |
J/(mol·K) | ||||
| Hfusion | 2 |
0.9080 |
1 |
4.1890 |
6.0050 |
5.1250 |
kJ/mol |
| Hvap | 2 |
2.3730 |
1 |
8.9720 |
13.7180 |
29.0180 |
kJ/mol |
| ηa | 2 |
548.2900 |
1 |
340.3500 |
1436.9300 |
||
| ηb | 2 |
−1.7190 |
1 |
−0.3500 |
−3.7880 |
||
| η | à T = 300 K |
0.0002942 |
Pa·s | ||||

Références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Joback method » (voir la liste des auteurs).
- Joback K. G., Reid R. C., "Estimation of Pure-Component Properties from Group-Contributions", Chem. Eng. Commun., 57, 233–243, 1987.
- Lydersen A. L., "Estimation of Critical Properties of Organic Compounds", University of Wisconsin College Engineering, Eng. Exp. Stn. Rep. 3, Madison, Wisconsin, 1955.
- Constantinou L., Gani R., "New Group Contribution Method for Estimating Properties of Pure Compounds", AIChE J., 40(10), 1697–1710, 1994.
- Nannoolal Y., Rarey J., Ramjugernath J., "Estimation of pure component properties Part 2. Estimation of critical property data by group contribution", Fluid Phase Equilib., 252(1–2), 1–27, 2007.
- Stein S. E., Brown R. L., "Estimation of Normal Boiling Points from Group Contributions", J. Chem. Inf. Comput. Sci. 34, 581–587 (1994).