Granulomatose éosinophilique avec polyangéite
La granulomatose éosinophilique avec polyangéite ou EGPA (en anglais Eosinophilic granulomatosis with polyangiitis), anciennement nommée syndrome de Churg-Strauss, est une forme de vascularite systémique[1] se référant à un groupe hétérogène de troubles caractérisés par la destruction inflammatoire de vaisseaux sanguins[2].
ou
granulomatose éosinophilique avec polyangéite
| Symptômes | Hémorragie et hémorragie digestive |
|---|
| Médicament | Mepolizumab |
|---|---|
| Spécialité | Immunologie et rhumatologie |
| CIM-10 | M30.1 |
|---|---|
| CIM-9 | 446.4 |
| DiseasesDB | 2685 |
| eMedicine |
1083013 derm/78 neuro/501 |
| MeSH | D015267 |
| Patient UK | Churg-strauss-syndrome |
![]() Mise en garde médicale
Mise en garde médicale
Les deux parties (artérielles et veineuses) du système sanguin peuvent être affectées.
Épidémiologie
La granulomatose éosinophilique avec polyangéite est très rare et peut affecter des sujets d’âges différents, principalement entre 40 et 45 ans. On distingue un ratio hommes-femmes égal à 1. En France, elle apparaît avec une fréquence de 2 à 7 personnes sur 10 millions d’adultes dans la population générale.
Mais cette prévalence semble augmenter à 64⁄100000 chez les patients asthmatiques traités, peu importe les médications utilisées, témoignant du terrain particulier sur lequel cette affection se développe[3].
Pathogénie
La pathogénie de la granulomatose éosinophilique avec polyangéite demeure énigmatique. Il s’agit très probablement d’une maladie auto-immune.
Néanmoins, il est fort probable que le premier événement pathogénique soit une réponse inflammatoire allergique après une exposition de l’appareil respiratoire à un antigène inhalé, car l’asthme est constitutif du syndrome. Il représente donc une manifestation clinique initiale dans la grande majorité des cas.
L’inflammation allergique joue un rôle important lors de la phase de vascularite, avec une augmentation de la production et une activation d’un type de globules blancs : les granulocytes éosinophiles.
Il existe une participation génétique. Ainsi, la maladie est plus fréquente en cas de groupe HLA-DRB4[4].
Le syndrome de Churg et Strauss est également une vascularite associée aux anticorps antineutrophiles cytoplasmatiques (p-ANCAs) dans environ la moitié des cas[5].
Anatomie pathologique
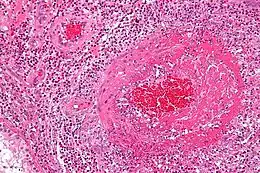
La granulomatose éosinophilique avec polyangéite est une vascularite nécrosante qui touche les artères et les veines de petit calibre avec lésions. C’est-à-dire que l’inflammation des vaisseaux entraîne l’épaississement et la fragilisation des parois. À l’inverse, une sténose peut se créer (rétrécissement des vaisseaux). Le flux sanguin en est donc perturbé, celui-ci ayant du mal à passer en quantité suffisante. Différents organes sont alors en ischémie (arrêt de passage du sang ). Cela peut être temporaire ou définitif, selon l’état de gravité du système atteint[6].
Comme souligné précédemment, différents organes peuvent aussi comporter un surnombre de polynucléaires éosinophiles. Ces globules blancs se rassemblent en granulomes et provoquent ainsi une réaction inflammatoire anormale. Les granulomes extravasculaires sont très caractéristiques de l’angéite (inflammation) de Churg et Strauss, mais ne sont ni constants, ni pathognomoniques.
Manifestations
Le syndrome de Churg et Strauss évolue habituellement en trois phases[5] :
- l’apparition d’un asthme rapidement cortico-dépendant chez trois quarts des patients ;
- l’hyperéosinophilie sanguine et tissulaire ;
- la vascularite systémique apparaissant en moyenne 9 ans après le début de l’asthme.
L’atteinte systémique s’installe rapidement, la gravité clinique est rapidement évidente, avec des signes généraux marqués [7] :
Manifestations pleuro-pulmonaires
Dans la majeure partie des cas, l’angéite systémique est précédée par un asthme fort et cortico-dépendant débutant relativement tard, aux alentours de la trentaine. Dans 38 à 70 % des cas, il comporte une anomalie de la radiographie pulmonaire. Il s’agit en général d’opacités alvéolaires localisées non-organisées dans les périphéries pulmonaires. le scanner thoracique retrouve les mêmes types d'anomalies à type d'opacités en verre dépoli, d'épaississement pleural, ou de nodules centrolobulaires[8].
Manifestations neurologiques
Les neuropathies périphériques sont les plus fréquentes et très suggestives du diagnostic. Aux membres inférieurs, l’atteinte la plus caractéristique est celle du nerf sciatique poplité externe chez 65,6 % des cas, et, à un moindre degré, du nerf sciatique poplité interne, tous deux localisés au niveau du genou.
Au niveau des membres supérieurs, il peut s’agir d’une atteinte du nerf médian. L’atteinte de plusieurs nerfs (multinévrite) est précoce au cours de l’évolution du syndrome de Churg et Strauss. Elle est sensitivomotrice, d’installation rapide, voire brutale.
La vascularite des petits vaisseaux crée des lésions ischémiques des nerfs périphériques. Les influx nerveux, aussi appelés potentiels d’action, moteurs et sensitifs sont alors absents ou diminués. Les neuropathies périphériques s’étendent par poussées successives, leur survenue est inopinée, et les manifestations allergiques précèdent souvent la venue du déficit moteur. Leur régression est lente et peut s’étendre de 12 à 18 mois, alors que les troubles moteurs ont totalement disparu.
Manifestations cutanées
On observe des manifestations cutanées diverses et variées assez fréquentes dans 40 à 70 % des cas, telles que des purpuras vasculaires, des nodules sous-cutanés rouges ou violacés au niveau des coudes, des doigts et du cuir chevelu.
Manifestations cardiaques
Les manifestations cardiaques apparaissent dans 35 % des cas, soit sous forme d’inflammation du péricarde ou d’inflammation myocardique
Manifestations oto-rhino-laryngées
Les manifestations oto-rhino-laryngées sont fréquentes sur 70 % des cas à la phase initiales de la maladie, à l’origine d’obstruction nasale, de sinusite et de polypose nasale. Une infection du sinus maxillaire est observée chez 62,5 % des patients.
Manifestations digestives
L’atteinte de l’appareil digestif est fréquente chez 30 à 60 % des cas. Les symptômes généralement observés sont les douleurs abdominales, et à un moindre degré les nausées, vomissements et diarrhée.
Examens et diagnostic
Les différents examens qui vont suivre permettent de diagnostiquer un SCS, mais généralement l’association d’un asthme, d’une altération importante du métabolisme général et d’une hyperéosinophilie doit faire évoquer le diagnostic du syndrome.
Biologie
Les analyses de sang vont mettre en évidence l’hyperéosinophilie en révélant un nombre anormalement élevé de polynucléaires éosinophiles. Elle va notamment montrer une augmentation de la vitesse de sédimentation chez 80 % des malades. Il peut exister une élévation des IgG4 ainsi que des anticorps antineutrophile cytoplasmatique (ANCA)[5]. Ces derniers, détectés en immunofluorescence, sont typiquement de localisation péri-nucléaires et sont souvent révélatrices de formes rénales ou pulmonaires[9]. le taux d'anticorps antimyéloperoxydase peut être augmenté[10].
La glomérulonéphrite peut être décelée par la présence de protéines ou de sang dans les urines.
Anatomopathologie
Des biopsies peuvent être faites au niveau de la peau, des muscles, des reins ou des poumons. L’observation au microscope montrera la présence de vascularite systémique et/ou d’éosinophiles en dehors des vaisseaux. Une inflammation des reins peut également être vue (glomérulonéphrite).
Imagerie
L’inflammation peut être détectée par une radiographie ou un scanner des poumons, en révélant la présence d’infiltrats pulmonaires et d’épaississement de la paroi des sinus.
Une IRM ou une échocardiographie peuvent compléter le diagnostic afin de visualiser le cœur et les vaisseaux.
Traitement
Avant l’introduction des corticoïdes et des immunosuppresseurs, l’issue du syndrome était presque toujours fatale. Sous traitement, le taux de survie dépasse cinq ans dans 90 %[11].
Corticoïdes
Au stade initial de la maladie, les corticoïdes, dont la prednisone et la prednisolone sont très efficaces, mais il faut les utiliser tout au long des différents stades de la maladie. En effet, ils permettent de contrôler la maladie rapidement grâce à leur effet anti-inflammatoire. On les administre principalement sous forme intraveineuse, la dose étant diminuée petit à petit jusqu’à obtenir une rémission complète[12]. Un traitement prolongé à petites doses est cependant nécessaire pour stabiliser l'asthme[13].
Immunosuppresseurs
Chez certains malades, le traitement par corticoïdes ne suffit pas. Dans ces cas, la prise d’un immunosuppresseur est nécessaire, afin de freiner l’activité du système immunitaire. Premièrement, le Cyclophosphamide permet de soulager l’inflammation au début du traitement. Puis le Méthotrexate et l’Azathioprine le remplacent au bout de 3 à 6 mois afin de maintenir la rémission. Le rituximab tend à devenir le traitement de références dans les formes résistantes aux corticoïdes[14].
Pronostic
Sous traitement, dans 90 % des cas, il y a une rémission complète de la vascularite. La survie à 10 ans s’élève à 79,4 %.
L’injection des corticoïdes permettent une amélioration rapide de l’état global du patient, et les douleurs et engourdissements dus à l’atteinte neurologique disparaissent progressivement.
Risques
Un quart des patients fait une rechute, et les manifestations associées sont majoritairement différentes et plus graves que celles observées initialement. Le risque de rechute est légèrement supérieur dans les formes ANCA positives[15].
Le traitement par corticoïdes doit durer un ou deux ans afin que la rémission soit stable, ce qui n’est pas à négliger sur l’influence de l’état psychologique du patient. Leur administration est responsable d’effets indésirables, comme l’hypertension, des troubles du sommeil et de l’humeur, des troubles hormonaux, une perte de masse musculaire, une ostéoporose (déminéralisation des os), des troubles digestifs, une prise de poids et un risque accru d’infection.
Les immunosuppresseurs augmentent le risque d’infection car le niveau de globules blancs baisse. Certains effets secondaires comme des saignements de la vessie, une chute de cheveux réversible, des nausées et une disparition de règles ont été observés.
Notes et références
- Merriam-Webster Définition du dictionnaire Merriam-Webster Online, consulté le 8 janvier 2009
- DermNet html Lexique des termes en dermatologie pathologique DermNet NZ ; consulté le 8 janvier 2009
- , Respir, “Syndrome de Churg et Strauss”
- Vaglio A, Buzio C, Zwerina J, Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art, Allergy, 2013;68:261-273
- Gioffredi A, Maritati F, Oliva E, Buzio C, Eosinophilic granulomatosis with polyangiitis: an overview, Front Immunol, 2014;5:549-549
- « Le syndrome de Churg et Strauss » sur Orphanet.
- François Lhote, Pascal Cohen, Philippe Guilpain, Loïc Guillevin, « Syndrome de Churg et Strauss » Revue du Praticien juin 2008. [PDF]
- Choi YH, Im JG, Han BK, Kim JH, Lee KY, Myoung NH, Thoracic manifestation of Churg-Strauss syndrome: radiologic and clinical findings, Chest, 2000;117:117-124
- Sinico RA, Di Toma L, Maggiore U et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome, Arthritis Rheum, 2005;52:2926-2935
- Choi HK, Liu S, Merkel PA, Colditz GA, Niles JL, Diagnostic performance of antineutrophil cytoplasmic antibody tests for idiopathic vasculitides: metaanalysis with a focus on antimyeloperoxidase antibodies, J Rheumatol, 2001;28:1584-1590
- Pagnoux C, Guilpain P, Guillevin L, Churg-Strauss syndrome, Curr Opin Rheumatol, 2007;19:25-32
- (en) Hellmich B, Gross WL, « Recent progress in the pharmacotherapy of Churg-Strauss syndrome », Expert Opin Pharmacother, vol. 5, no 1, , p. 25-35 (PMID 14680433, DOI 10.1517/14656566.5.1.25)
- Sinico RA, Bottero P, Churg-Strauss angiitis, Best Pract Res Clin Rheumatol, 2009;23:355-366
- Mohammad AJ, Hot A, Arndt F et al. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss), Ann Rheum Dis, 2016;75:396-401
- Comarmond C, Pagnoux C, Khellaf M et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort, Arthritis Rheum, 2013;65:270-281
Liens externes
- Site officiel de l'association de patients France Vascularites
- « Portail des maladies rares et des médicaments orphelins », sur Orphanet
- Syndrome de Churg-Strauss (CSS) - Granulomatose éosinophile avec polyangéite (EGPA) - Pour la langue française, cliquez sur le bouton TRANSLATE puis sur le drapeau
- Protocole National de Diagnostic et de Soins PNDS