Fonction d'état
Une fonction d'état est une fonction de grandeurs appelées variables d'état, qui définissent l'état d'équilibre d'un système thermodynamique. Les variables d'état sont par exemple la température , la pression , le volume ou le nombre de moles . Une telle fonction possède donc la propriété de ne dépendre que de l'état d'équilibre dans lequel se trouve le système, quel que soit le chemin emprunté par le système pour arriver à cet état. En particulier, au cours d'une transformation entre deux états d'équilibre, la variation d'une fonction d'état ne dépend pas du chemin suivi par le système pendant la transformation, mais uniquement des états d'équilibre initial et final. Ceci permet de déterminer cette variation pour n'importe quelle transformation, y compris une transformation irréversible, en imaginant un chemin réversible pour lequel le calcul est possible.




L’énergie interne et l’entropie sont des exemples de fonctions d'état. D'une façon générale, les potentiels thermodynamiques sont également des fonctions d'état particulièrement importantes en physique et en chimie.


Quant au travail et à la chaleur échangés pendant une transformation, ils ne peuvent pas être assimilés à la variation d'une fonction d'état car ils dépendent de la nature de la transformation. Au cas général, le travail et la chaleur sont plutôt les fonctions de parcours parce que leurs valeurs dépendent du chemin parcouru entre l'état initial et l'état final du système. Néanmoins, il existe des cas particuliers où la chaleur et le travail ne dépendent plus du chemin suivi, qu'il soit réversible ou irréversible, lorsque les transformations s’effectuent soit à pression constante (voir enthalpie) ou à volume constant.
Mathématiquement, la possibilité de représenter une grandeur physique par une fonction d'état est liée aux propriétés de sa forme différentielle, c'est-à-dire sa variation dans une transformation infinitésimale autour d'un état d'équilibre.
Exemple de compréhension
Considérons la fonction altitude A lors d’une randonnée en montagne. L'« état » du groupe de randonneurs peut être défini par exemple, par ses coordonnées GPS permettant de le situer sur le chemin de randonnée. Supposons que pour aller d’un sommet (1) à 2 500 m à un sommet (2) à 2 600 m, deux chemins s’offrent au groupe :
- un premier chemin qui suit la ligne de crête presque au niveau des deux sommets ;
- un deuxième chemin qui redescend dans la vallée à 500 m d’altitude.
La variation d’altitude ΔA est la même pour les deux chemins :
- ∆A = A(2) – A(1) = 100 m
La fonction altitude pourrait être considérée comme une fonction d’état de la randonnée. En revanche, les efforts consentis, travail et chaleur dégagée par les randonneurs ne seront pas identiques ! Ces grandeurs ne seraient donc pas des fonctions d’état mais des grandeurs liées au chemin suivi.
Variables d’état
Certaines fonctions d’état jouent un rôle particulier dans la définition des états d’équilibre d’un système. Ce sont des grandeurs accessibles, à l’échelle macroscopique, directement ou indirectement grâce à des instruments de mesure :
- la pression p exprimée en Pa (pascal) ;
- la température T exprimée en K (kelvin) ;
- le volume V exprimé en m3 (mètre cube) ;
- la quantité de matière n exprimée en mol (mole).
Ces fonctions d’état particulières sont appelées variables d’état d’équilibre d’un système thermodynamique.
Certaines de ces variables d’état sont des grandeurs intensives, comme la température et la pression. Cela signifie qu’elles ne dépendent pas de l'extension du système, en d'autres termes de la quantité de matière du système.
Exemple : si on mélange deux bouteilles contenant 1 litre d’eau chacune, à la température de 20 °C, la température finale est 20 °C et non pas 40 °C. Il en serait de même avec la pression qui ne présente pas non plus la propriété d’additivité. En revanche, le volume V final sera égal à 2 l. Le volume n’est pas une grandeur intensive mais une grandeur extensive qui dépend de l'extension du système et donc de la quantité de matière. La quantité de matière n, possède elle-même cette propriété d'additivité et est donc également une grandeur extensive.
Les variables intensives sont importantes pour définir l'état d'équilibre d'un système physico-chimique. En effet l'équilibre est atteint lorsque la valeur des variables intensives est homogène dans tout le système et ne varie pas au cours du temps.
Remarque: Dans le cas des systèmes chimiques comportant plusieurs espèces chimiques en réaction, une autre variable doit être prise en compte. Il s'agit de la variable de composition ξ qui permet de déterminer la composition du système chimique pour un avancement donné de la réaction.
Équation d’état
Les variables d’état définissant l’état d’équilibre d’un système, p, V, T, n, ne sont pas indépendantes. Elles sont liées par une relation appelée équation d’état du système, plus ou moins complexe.
Par exemple, l’équation d’état la plus simple est celle du gaz parfait. C'est le modèle idéalisé d'un gaz constitué de particules suffisamment éloignées les unes des autres pour considérer qu'il n'y a aucune interaction d'ordre électrostatique entre elles ; cela implique que la pression est faible. Dans ces conditions, l'équation d'état est indépendante de la nature chimique du gaz considéré comme parfait. De nombreux gaz réels dans les conditions normales de température et de pression vérifient, avec une excellente approximation, le modèle du gaz parfait ; c'est le cas des gaz constituants de l'air : le diazote (N2) et le dioxygène (O2).
Elle s'exprime par la relation :
- , où R est la constante des gaz parfaits, R = 8,314 J.K−1 .mol−1)

En mesurant T et p pour n (mol.) de gaz parfait on peut alors calculer le volume et définir parfaitement son état d'équilibre :

Pour définir l’état d’une quantité donnée d'un gaz parfait (n fixé), 2 variables indépendantes suffisent (cette propriété peut être étendue à tous les corps purs, qu'ils soient solides, liquides ou gazeux).
Propriété fondamentale des fonctions d'état
Rappel de définitions mathématiques
La différentielle d'une fonction d'état, fonction de plusieurs variables indépendantes, est une différentielle totale exacte. Cela signifie qu'elle est égale à la somme de ses différentielles partielles par rapport à chaque variable. Pour une fonction de deux variables notée :


est la dérivée partielle de F par rapport à x et de même pour y.

Application : si F est fonction de plusieurs variables au cours d'une transformation, on peut décomposer cette transformation en plusieurs étapes de telle manière que pour chaque étape une seule variable indépendante varie, ce qui rend l'étude plus simple. La variation globale de F sera égale à la somme des variations partielles de chaque étape et sera bien évidemment identique à la variation obtenue au cours de la transformation effectuée en une seule étape; toutes les variables variant simultanément.
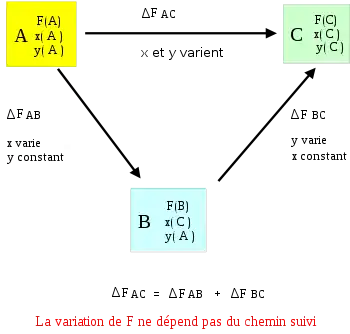
Considérons une transformation définie par l'état initial A : F(A) ; x(A) ; y(A) et l'état final C : F(C) ; x(C) ; y(C).
On définit un état intermédiaire B : F(B) ; x(B) = x(C) ; y(B) = y(A).
On dit alors que la variation de la fonction d'état ne dépend pas du chemin suivi.
Calculons la variation de la fonction :


Remarque : L'ordre de variation des variables indépendantes x et y n'a aucune incidence sur le résultat. Cela se traduit mathématiquement par le fait que les dérivées secondes croisées de la fonction F par rapport à x et y sont égales.

Exemple d'application : cas du gaz parfait
On considère un gaz parfait dont l'état est donné par sa température T et sa pression p. Pour effectuer une transformation, on fait varier la pression ou la température du gaz de valeurs initiales notées et à des valeurs finales et . La quantité de matière est considérée comme constante.




Cas du volume
L'équation d'état du gaz parfait nous donne une expression explicite du volume en fonction de ces deux paramètres :
.

Le volume apparaît donc comme une fonction d'état. On peut calculer à partir de cette expression la variation de volume du gaz pendant la transformation de l'état initial vers l'état final :
![\Delta V=V_{f}-V_{i}=nR\left[{\frac {\ T_{f}}{\ p_{f}}}-{\frac {\ T_{i}}{\ p_{i}}}\right]](https://img.franco.wiki/i/7aaf96f33dc3bc73ce1fbf1c9fda61242695da7b.svg)
On peut vérifier qu'on retrouve la même variation en suivant deux chemins différents pour la transformation. Pour le premier chemin, on fait varier la pression de à en maintenant la température constante et égale à , puis on fait varier la température de à en maintenant la pression constante et égale à puis l'inverse. Dans le second chemin, on procède de la même manière, mais en faisant varier la température avant la pression.
Pour une petite variation infinitésimale de la pression et de la température, on peut écrire :

soit, en utilisant l'équation d'état :

On considère les deux chemins différents définis par l'ordre de variation de p et T
- Pour le premier chemin, on peut écrire la variation totale de volume comme suit en faisant varier d'abord la pression :

ce qui donne


![\Delta V_{1}=V_{f}-V_{i}=nR\left[{\frac {\ T_{f}}{\ p_{f}}}-{\frac {\ T_{i}}{\ p_{i}}}\right]](https://img.franco.wiki/i/fecedc1b3b791f280996063dd327438e4d11e251.svg)
- Pour le second chemin, on fait varier la température en premier :

On retrouve le même résultat que pour le premier chemin, qui est évidemment le même que celui obtenu directement à partir de l'équation d'état.
Pour que la forme différentielle dV de la fonction V(T, p) soit une différentielle exacte, il faut que l'ordre de la dérivation de V par rapport à T et p soit indifférent ou encore que les dérivées secondes croisées soient égales, ce qui est le cas :

Cas du travail
En revanche, le travail des forces de pression dépend du chemin suivi, il ne peut donc pas être écrit comme la variation d'une fonction d'état, et le travail fourni au cours d'une transformation infinitésimale n'est pas une différentielle exacte.

Nous avons établi précédemment la différentielle du volume d'un gaz parfait :
or le travail des forces de pression

donc la forme différentielle du travail associé à un gaz parfait, est égale à :

On peut démontrer que n'est pas une différentielle exacte en remarquant que les dérivées secondes croisées ne sont pas égales.

Ceci est confirmé par le calcul du travail fourni au cours de la transformation en suivant les deux chemins différents décrits précédemment. On trouve alors pour le premier chemin un travail fourni


et pour le second

Le travail total obtenu dans les deux cas envisagés est différent. Il dépend donc du chemin suivi et ne peut pas être écrit sous la forme de la variation d'une fonction d'état.
Remarque :
- Si la pression est constante, le travail mis en jeu ne dépend plus du chemin suivi puisqu'il ne dépend plus que de la variation d'une variable d'état.


Cette propriété est similaire à celle concernant la chaleur mise en jeu à pression constante égale à une variation d'enthalpie (ΔH = Qp) ou à volume constant, égale à une variation d'énergie interne (ΔU = Qv). Dans ces deux cas, la chaleur ne dépend plus du chemin suivi puisqu'elle est égale à la variation d'une fonction d'état. Précisons qu'à volume constant le travail des forces de pression est nul.
Intérêt de la propriété des fonctions d'état en thermodynamique
Les transformations réelles sont irréversibles et leur déroulement dépend de la façon de procéder. Elles ne sont donc pas modélisables mathématiquement et le calcul des grandeurs thermodynamiques qui leur sont associées, est impossible. Néanmoins, si cette grandeur est une fonction d'état, sa variation ne dépend que de l'état final et de l'état initial d'équilibre. Pour calculer cette variation il suffit alors d'imaginer une transformation réversible, partant du même état initial pour aboutir au même état final que pour la transformation réelle. Cette transformation réversible est caractérisée par une succession d'états d'équilibres. Elle est modélisable mathématiquement et sa variation est donc calculable.
Cette variation est identique à celle observée pour la transformation irréversible et le problème est résolu.
En outre si la fonction d'état est fonction de plusieurs variables, on pourra décomposer la transformation en autant d'étapes intermédiaires réversibles qu'il y a de variables; chaque étape étant caractérisée par la variation d'une seule variable indépendante. Cela simplifie grandement les calculs.
Fonctions d’état usuelles en thermodynamique
- L’énergie interne : U exprimée en J (joule)
- L’enthalpie : H = U + pV exprimée en J
- L’entropie : S exprimée en J.K-1
- L’énergie libre : F = U – T.S exprimée en J
- L’enthalpie libre : G = H – T.S exprimée en J
Bibliographie
- (en) Walter Greiner, Ludwig Neise; et Horst Stöcker, Thermodynamics and statistical mechanics, New York, Springer-Verlag, coll. « Classical theoretical physics », , 463 p. (ISBN 978-0-387-94299-5 et 978-3-540-94299-3, OCLC 30814519, lire en ligne), p. 25-32
- Paul Arnaud, Cours de chimie physique, Paris, Dunod, , 529 p. (ISBN 978-2-10-001640-2, OCLC 2100016407).
- Peter Atkins et Julio De Paula (trad. de l'anglais par Jean Toullec et Monique Mottet), Chimie Physique, Bruxelles, De Boeck, , 4e éd., 973 p. (ISBN 978-2-8041-6651-9, BNF 43642948, lire en ligne), p. 47.