Dégénérescence corticobasale
La dégénérescence corticobasale (DCB) ou dégénération corticobasale ganglionnaire est une maladie neurodégénérative relativement rare mais de plus en plus fréquemment décrite, touchant principalement les régions cérébrales sous-corticales, puis corticales et les noyaux gris centraux et présentant typiquement un dysfonctionnement moteur asymétrique ainsi que des troubles cognitifs[1]. Le diagnostic clinique est difficile à cause de la variabilité des symptômes.
| CIM-9 | 331.6 |
|---|---|
| DiseasesDB | 33284 |
| eMedicine | 1150039 |
| MeSH | D000088282 |
![]() Mise en garde médicale
Mise en garde médicale
Le médecin peut s'aider de l'imagerie.
Le traitement est encore très limité.
Diagnostic
Historique
La dégénérescence corticobasale a été décrite pour la première fois en 1967-1968 par Rebeiz, Kolodny et Richardson, qui rapportent un syndrome comprenant[2] - [3] :
- une rigidité
- une akinésie asymétrique,
- une dystonie des membres supérieurs,
- une apraxie,
- des myoclonies
- une démence
Bien que reconnue comme une maladie distincte, la DCB est, aussi, souvent considérée comme l'une des variantes de la PSP (paralysie supranucléaire progressive) au même titre que le syndrome de Guam est une variante de la PSP. La DCB comme la PSP est classée dans le groupe des syndromes parkinsoniens atypiques dits "Parkinson Plus".
Présentation clinique
Au début de la maladie, classiquement, on retrouve un dysfonctionnement moteur asymétrique au niveau des membres supérieurs, une perte sensorielle corticale et une apraxie sans détérioration intellectuelle. Le tableau clinique peut souvent ressembler à celui d'une paralysie supranucléaire progressive (PSP), avec une résistance à la L-DOPA, un tremblement d'attitude, un syndrome pseudobulbaire avec dysarthrie, dysphagie, troubles oculomoteurs ainsi qu'une démence. La détérioration intellectuelle s'installe ensuite, elle est liée à l'atteinte du cortex associatif, et notamment du cortex frontal[4].
Certains auteurs[5] ont proposé comme critère diagnostic la présence de 3 des symptômes suivants :
- Bradykinésie[6] et rigidité résistante à la L-DOPA
- Apraxie idéomotrice asymétrique ou syndrome du membre étranger
- Signes sensoriels corticaux
- Dystonie focale aux membres
- Tremblement d'attention
- Myoclonie
Les critères d'exclusion (leur présence exclut le diagnostic) sont[7] : une bonne réponse au L-DOPA (comme dans la maladie de Parkinson), un tremblement de repos, des atteintes précoces du regard vertical (comme dans la paralysie supranucléaire progressive), des atteintes sévères du système nerveux autonome (comme dans l'atrophie multisystémique), et toutes anomalies aux examens d'imagerie qui pourraient expliquer les symptômes.
À noter que la démence précoce est désormais reconnue comme l'un des symptômes initiaux de la dégénérescence corticobasale.
Diagnostic différentiel

Le diagnostic est difficile, surtout au stade initial : même des cliniciens expérimentés se trompent, en 1997, dans plus de 50 % des cas[8]. La dégénérescence corticobasale peut être difficile à différencier de la PSP ou d'une atrophie multisystémique. Lorsque les signes corticaux apparaissent, le diagnostic est facilité. Les diagnostics différentiels principaux sont :
- maladie d'Alzheimer (MA)
- maladie de Parkinson (MP)
- paralysie supranucléaire progressive (PSP ou maladie de Steele-Richardson-Olszewski)
- dégénérescence frontotemporale (DFT) dont la maladie de Pick et la FTDP-17
- atrophie multi-systématisée (AMS)
- Intoxication chronique aux Annonaceae: une des deux formes de dégénérescence cérébrale induite par ces fruits (Gd-PDC: syndrome parkinsonien et démentiel guadeloupéen) se manifeste par une atrophie des lobes frontal et pariétal[9] ainsi que de la substantia nigra[10].
Imagerie
La scintigraphie cérébrale montre un hypodébit pariétofrontal asymétrique. L'IRM montre une atrophie pariétale et frontale asymétrique et un élargissement des ventricules latéraux en regard de l'atrophie.
Anatomopathologie
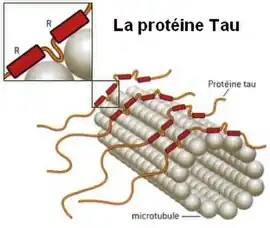
Il y a une dégénérescence neurofibrillaire dans les noyaux sous-corticaux et dans le cortex, surtout dans les régions frontales. On retrouve une perte neuronale, une gliose et une accumulation de protéine tau dans les astrocytes. On note aussi la présence de cellules chromatolytiques (ou cellules de Pick). Les zones les plus touchées sont les régions frontales et pariétales.
Examen neuropathologique post-mortem
Comme pour les autres maladies neurodégénératives, il reste, à ce jour, le seul examen permettant de porter un diagnostic définitif de dégénérescence corticobasale. À cet égard, les patients et leur famille qui font don de leur cerveau[11] permettent des avancées considérables pour connaître et lutter contre les DCB et les maladies neurodégénératives.
Physiopathologie
La ou les cause(s) sont inconnues. Il semble que cette maladie serait la conséquence d'une dégénérescence des noyaux gris centraux, spécifiquement marqués par la dégénérescence neuronale ou la dépigmentation (perte de mélanine dans le neurone) dans la substance noire[12]. On décèle aussi une atrophie asymétrique des régions corticales postérieures et pariétales[13]. Des études post mortem des patients diagnostiqués avec une dégénérescence corticobasale révèlent un ballonnement des neurones, une gliose, une tauopathie[12].
Évolution
Le début de la maladie se situe entre 60 et 70 ans. Le plus jeune cas décrit est celui d'une femme de 28 ans[14].
(NB : ce type de moyenne n'est pas très significatif car les écarts entre les espérances de vie les plus courtes et les plus longues, jusqu'à 15 ans et plus, sont très variables en fonction de l'état général du malade, de son âge, etc.)
Épidémiologie
La présentation clinique de la dégénérescence corticobasale n'apparaît souvent pas avant l'âge de 60 ans. Les diagnostics faits le plus précocement ont été faits à 28 ans[15]. Bien que la DCB soit présente chez les hommes et chez les femmes, il existe une prédominance féminine. Des calculs récents suggèrent que la prévalence de la DCB est de 4,9 à 7,3 pour 100 000 personnes. Le pronostic d'un individu diagnostiqué avec une DCB est d'environ 8 ans, bien que certains patients aient vécu plus de 13 ans et aient toujours un état correct, mais avec des troubles sérieux comme une dysphagie, et une rigidité des membres générale. L'utilisation partielle de sonde nasogastrique peut être nécessaire et aidera à prévenir les pneumopathies d'inhalation, la première cause de décès dans la DCB. L'incontinence est fréquente car les patients ne peuvent souvent pas exprimer leur besoin d'aller aux toilettes, du fait de troubles de la parole. Par conséquent, une hygiène soigneuse est nécessaire pour prévenir les infections urinaires [16]
Prise en charge

Biologique
La L-Dopa, traitement de référence pour la maladie de Parkinson, n'est pas efficace sur cette maladie qui fait partie des syndromes parkinsoniens et il n'y a pas, à ce jour, de traitement spécifique permettant de ralentir l'évolution de la maladie.
Depuis mars 2009, la DCB a un centre d'expertise (Centre de Référence) dans le cadre de la mise en place du Réseau National des Centres de Référence et de Compétence pour les Démences Rares[17].
Prise en charge familiale et environnementale
À la demande de familles de malades DCB et en raison de la proximité des maladies que sont la DCB et la PSP, l'association PSP-France a pris en charge le soutien aux familles dont l'un des proches serait atteint par la DCB.
Références
- « Corticobasal Degeneration Information Page: National Institute of Neurological Disorders and Stroke (NINDS) » (consulté le ).
- (en) Rebeiz JJ, Kolodny EH, Richardson EP Jr. « Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life » Trans Am Neurol Assoc. 1967;92:23–26. .
- (en) Rebeiz JJ, Kolodny EH, Richardson EP Jr. « Corticodentatonigral degeneration with neuronal achromasia » Arch Neurol. 1968 Jan;18(1):20–33.
- Dégénérescence cortico-basale sur http://www.orpha.net.
- Riley DE, Lange AE, Lewis A, et al. Cortico-basal ganglionic degeneration. Neurology 1990;40:1203-1212.
- La bradykinésie est un trouble moteur, caractérisé par un ralentissement des mouvements et la perte de leur finesse d'exécution, comme ceux de l'écriture. Ce trouble moteur est un des symptômes de la maladie de Parkinson.
- Farsang M et al. Clinical features of corticobasal degeneration. Ideggyogy Sz. 2005 Jan 20;58(1-2):45-51. .
- (en) Litvan I, Agid Y, Goetz C, Jankovic J, Wenning GK, Brandel JP, Lai EC, Verny M, Ray-Chaudhuri K, McKee A, Jellinger K, Pearce RK, Bartko JJ, « Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study », Neurology, vol. 48, no 1, , p. 119-25. (PMID 9008506).
- « Fig. 1 Mesencephalic atrophy in Gd-PSP. Sagittal T 1 -weighted sequence... », sur ResearchGate (consulté le ).
- Lannuzel, Annie, Höglinger, G. U. et Verhaeghe, S., « Atypical parkinsonism in Guadeloupe : a common risk factor for two closely related phenotypes? », sur oup.com, (consulté le ).
- « Don du cerveau pour la recherche », sur neuroceb.org (consulté le ).
- (en) Scaravilli T, Tolosa E, Ferrer I. « Progressive supranuclear palsy and corticobasal degeneration: Lumping versus splitting » Movement Disorders 2005;20:S21-S8.
- (en) Wadia PM, Lang AE, « The many faces of corticobasal degeneration », Parkinsonism Relat Disord, vol. 13 Suppl 3, , S336-40. (PMID 18267261, DOI 10.1016/S1353-8020(08)70027-0).
- DePold Hohler A et al. The youngest reported case of corticobasal degeneration. Parkinsonism Relat Disord. 2003 Oct;10(1):47-50. .
- Seritan AL, Mendez MF, Silverman DHS, Hurley RA, Taber KH. 2004. Functional imaging as a window to dementia: Corticobasal degeneration. Journal of Neuropsychiatry and Clinical Neurosciences 16:393-9.
- Mahapatra RK, Edwards MJ, Schott JM, Bhatia KP. 2004. Corticobasal degeneration. Lancet Neurology 3:736-43.
- site internet du Centre de Référence pour les démences rares.
Liens externes
- Dégénérescence corticobasale sur http://www.orpha.net
- Dégénérescence cortico-basale sur http://www.cref-demrares.fr/
- traduction automatique de Dégénérescence cortico-basale de la Wikipédia en anglais
Pour aller plus loin
- Walusinski O. Gilles de la Tourette, beyond the eponym. Oxford University Press. 2019.
- Rickards H, Cavanna AE. Gilles de la Tourette: the man behind the syndrome. J Psychosom 2009;67:4669-74.