Syndrome de Rett
Le syndrome de Rett est une maladie génétique rare se développant chez le très jeune enfant, principalement la fille, et provoquant un handicap mental et des atteintes motrices sévères.

| Spécialité | Pédiatrie, psychiatrie et neurologie |
|---|
| CIM-10 | F84.2 |
|---|---|
| CIM-9 | 330.8 |
| OMIM | 312750 |
| DiseasesDB | 29908 |
| MedlinePlus | 001536 |
| eMedicine | 916377 |
| MeSH | D015518 |
| MeSH | C10.574.500.775 |
| GeneReviews |
![]() Mise en garde médicale
Mise en garde médicale
Il s'agit de la première cause de polyhandicap d'origine génétique chez les filles et on estime qu'il touche environ 50 nouvelles personnes par an en France.
Génétique
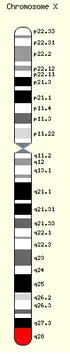
Le syndrome de Rett typique est associé dans près de 95 à 97 % des cas à une mutation dans le gène MECP2 (Méthyl-CpG-binding protein 2), localisé sur le bras long (q) du chromosome X, dans la région Xq28[1]. Il s'agit d'une encéphalopathie neurodéveloppementale très particulière, touchant essentiellement les filles, et étant dans plus de 99 % des cas une mutation de novo.
Dans sa forme typique, elle est caractérisée par une décélération globale du développement psychomoteur, puis par une perte des acquisitions cognitives et motrices, survenant après une période de développement normal.
Dans les formes atypiques, des mutations dans le gène CDKL5 (Cyclin dependent kinaselike 5) et dans le gène FOXG1 (Forkhead box G1) ont été retrouvées chez des enfants avec épilepsie précoce ou porteurs de formes congénitales. Certains patients diagnostiqués en raison de leur phénotype, mais négatifs pour les mutations dans les gènes MECP2, CDKL5 ou FOXG1, ont été soumis au séquençage complet de l'exome, ce qui a permis d'identifier plusieurs dizaines de nouveaux gènes[2], tels que WDR45[3], GRIN2B[4], STXBP1, GABRB2 et GABRG2[5], KIF1A[6].
Description
Le syndrome de Rett commence à se manifester chez l'enfant 6 à 18 mois après une période d'évolution normale.
Une période de régression apparaît alors avec[7] :
- perte partielle ou complète de l'utilisation volontaire des mains ; avec apparition de stéréotypies manuelles caractéristiques à tous les stades de la maladie (pressions, torsion, applaudissement, frottement, lavage des mains) ; Selon Einspieler et al. en 2005, et Temudo et al. en 2007 : ces stéréotypies de la main coïncident avec la perte de contrôle volontaires des mouvements de la main en début du développement de la maladie, voire la précèdent[8] - [9].
- perte partielle ou complète du langage ;
- perte ou défaut d'acquisition de la marche ou marche « robotisée » ;
- dysfonctionnement respiratoire avec des épisodes d'apnée pendant l'éveil, d'hyperventilation intermittente, des épisodes de blocage de la respiration, d'expulsion de l'air ou de salive ;
- troubles de la communication et retrait social dès la petite enfance ;
- détérioration du comportement, manifestations autistiques et rires ou excès de cris ou de pleurs inappropriés ;
- apparence de retard mental sévère, due à des tests de QI non adaptés[10] - [11] - [12] ;
- crises d'épilepsie ;
- déformations orthopédiques (scoliose, cyphose) ;
- communication par le regard intense – « eye pointing » ;
- ostéoporose ;
- troubles du tonus ;
- troubles du sommeil ;
- retard de croissance ;
- troubles vasomoteurs ;
- mains et pieds petits, hypotrophiques ;
- bruxisme pendant l'éveil.
Ni les patientes atteintes du syndrome de Rett, ni les souris modèle de la pathologie ne présentent de signes de dégénérescence cellulaire. Ceci suggère que la maladie est provoquée par un dysfonctionnement des réseaux neuronaux plutôt que par une perte de neurones. Grâce à des travaux menés chez des souris modèles, l’équipe d’Adrian Bird, en Écosse, a montré en 2007[13] que ce syndrome était réversible si la protéine Mecp2 était ré-exprimée, même à un stade avancé de la maladie[14].
Prévalence
Le syndrome de Rett représente l'une des principales causes de retard mental chez la femme dans le monde, sa prévalence est selon le CNRS de 1⁄10 000 enfants[15]. Il s'agit de la première cause de polyhandicap d'origine génétique en France chez les filles[16].
Critères de diagnostic
Clinique
Les critères de diagnostic du syndrome de Rett, retenus en 1988 et toujours d'actualité, sont[17] :
- un développement apparemment normal jusqu’à l’âge de 6 à 18 mois ;
- un périmètre crânien normal à la naissance, ralentissement de la croissance du crâne entre 3 mois et 4 ans ;
- l'absence d’un développement normal du langage ;
- la présence de mouvements répétitifs des mains (lavage de mains, torsions, etc.) ;
- une apraxie/ataxie du tronc et perte de l’utilisation volontaire des mains entre six et trente mois ;
- une démarche instable ou mal assurée (lorsque l’enfant marche).
Les critères d'exclusion qui, s'ils sont présents, permettent d'exclure la maladie de Rett sont :
- une rétinopathie ou atrophie optique ;
- un retard de développement intra-utérin ;
- une microcéphalie congénitale ;
- des signes d'une maladie métabolique identifiable ;
- des signes d'une maladie neurologique identifiable et progressive ;
- des lésions cérébrales acquises en période périnatale (à la suite d'une infection ou d'un traumatisme crânien) ;
- une viscéromégalie ou des signes d'une maladie de surcharge.
D'autres symptômes peuvent être présents et aider au diagnostic : épilepsie, trouble du sommeil, anomalies à l'électroencéphalogramme, spasticité musculaire avec atrophie musculaire et dystonie, scoliose, hypotrophie des pieds, raccourcissement du 4e métacarpien et/ou métatarsien à la radiographie, retard de croissance, grincements de dents, trouble de la déglutition et de la mastication, mauvaise circulation des membres inférieurs.
Les troubles respiratoires sont fréquents et peuvent être de plusieurs types, nécessitant un prise en charge différente[18] : hyperventilation avec un risque d'alcalose respiratoire ou, au contraire, hypoventilation avec acidose respiratoire, pouvant être accompagnée d’apnées.
Les autres symptômes comportementaux sont l'irritabilité, l'agitation avec hurlements et peur des agressions, des pleurs inconsolables, un regard fuyant (évitant de croiser le regard des autres). Il peut exister une absence d’implication émotionnelle ou sociale, avec absence de sujets d’intérêt en général et un refus marqué d'utiliser les comportements sociaux non verbaux, ce qui peut évoquer un autisme.
Diagnostic différentiel
Évolution
Après une période silencieuse, pendant laquelle le développement est normal, le syndrome de Rett évolue en 4 stades[19] :
- Le stade I de stagnation précoce, entre 6 et 18 mois, avec retard des acquisitions psychomotrices sans réelle régression ;
- Le stade II de régression neurologique rapide, entre 1 et 4 ans, avec régression des acquisitions motrices et mentales, perte de l'usage des mains et apparition de stéréotypies manuelles typiques ;
- Le stade III de stabilisation apparente, dite « phase de réveil », entre 2 et 10 ans, concerne essentiellement les filles ayant acquis la marche. Il y a régression motrice mais amélioration des capacités de communication avec contact visuel et diminution des caractères autistiques. La majorité des patientes gardent une « pseudo marche » jusqu'à l'âge adulte ;
- Le stade IV de détérioration motrice tardive, après 10 ans. Avec perte de la marche si elle avait été acquise, trouble du tonus et déformation squelettique. Malgré cette détérioration motrice, la socialisation et le contact visuel sont conservés pendant toute la vie adulte.
Pronostic
Le syndrome de Rett n'est identifié que depuis 1983. Encore aujourd'hui, de nombreuses personnes adultes atteintes du syndrome de Rett ne sont pas ou sont mal diagnostiquées. Dans ces conditions et s'agissant d'une maladie rare, il est difficile d'établir une analyse statistique fiable.
La survie est relativement bonne et les patientes dépassent habituellement l'âge de 10 ans et 70 % d'entre elles atteignent les 35 ans. Cette survie prolongée implique de prévoir une prise en charge multi-disciplinaire sur le long terme.
Il existe une fréquence élevée de mort subite inexpliquée à l'âge adulte.
On connait[20] cependant le cas d'une Norvégienne qui a atteint l'âge de 60 ans (mais aucun test génétique n'a permis de confirmer le diagnostic) et d'une Danoise, née en 1923, chez qui on a retrouvé une anomalie du gène lorsqu'elle a eu 66 ans ; elle est décédée à l'âge de 79 ans, des suites d'une péritonite[21]. Ann-Charlotte “Lottan” Holmström, née en 1948 en Suède, est certainement l'une des personnes les plus âgées, touchée par la maladie[22].
Traitement
Pour l'humain, il n'existe pas à ce jour de traitement curatif pour les enfants atteints. Un premier médicament est cependant disponible aux USA depuis 2023.
Les traitements symptomatiques doivent être entrepris dès que nécessaire avec par exemple un traitement anti-épileptique si nécessaire, une prise en charge médicale en cas de scoliose sévère, une alimentation adaptée et riche en apport calcique (l'ostéoporose étant parfois aussi traitée par l'acide zolédronique[23]).
La prise en charge éducative est particulièrement importante et doit être adaptée au cas par cas. Cette prise en charge doit être entreprise le plus tôt possible pour avoir le plus de chance de progression. Pour essayer d'obtenir la plus grande autonomie possible de la part de la patiente, il faut travailler et entretenir sa motricité et sa coordination avec un kinésithérapeute, un ergothérapeute et un psychomotricien, un orthophoniste[24], etc. ; proposer des activités variées, sportives et ludiques, pour faciliter son intégration à l'environnement ; encourager et développer ses facultés relationnelles de communication et d’échange.
En matière de communication, l'exemple de Karly Wahlin (1985-2012) est un espoir pour les malades et leurs familles. Atteinte du syndrome de Rett, la jeune Américaine avait à l'âge de 10 ans appris à utiliser un clavier d'ordinateur pour communiquer. Elle écrivait des poèmes, composait de la musique, et tenait même un blog[25].
En France, les parents peuvent recourir à l’Association française du syndrome de Rett[26].
Un projet dit GazePlay qui est un logiciel libre (et gratuit) regroupant (en 2020) une soixantaine de mini-jeux jouables avec un eye-tracker ou oculomètre (système de contrôle par l'œil du curseur de la souris d'un ordinateur), associé à une communauté pour les personnes en situation de polyhandicap, avec interface disponible (en 2020) en langues française, anglaise, allemande et néerlandaise et « partiellement traduits dans une vingtaine d’autres langues »[27]. Certains jeux et/ou outils utilisent le regard ou un oculomètre (ex : Eye Tribe ou Tobii 4C) pour proposer des solutions encore plus efficaces, telles que par exemple créées par Townend [28] - [29], avec une CAA (communication augmentée et alternative) basée sur le regard, conçu avec deux jeunes filles atteintes du syndrome de Rett.
Médicaments
Le 11 mars 2023, la FDA a autorisé la mise sur le marché de la molécule appelée trofinetide sous la dénomination commerciale Daybue[30]. Le médicament devait être disponible à partir de la fin du mois d'avril 2023 aux USA pour les malades âgés de plus de 2 ans. Selon Acadia, le trofinetide permet de diminuer certains symptômes handicapants de la maladie, sans toutefois les supprimer complètement.
Recherche
Médicaments
L'efficacité d'un traitement in vivo par la cystéamine a été démontrée [31]. Cette molécule agit sur les mécanismes du transport axonal. Les souris traitées ont une durée de vie augmentée et une fonction motrice améliorée par rapport aux animaux non traités. La cystéamine est autorisée aux États-Unis dans le traitement d'une autre maladie rare de l'enfant, la cystinose.
Il a été démontré que les récepteurs d'une substance chimique du cerveau appelée glutamate, en particulier le type NMDA, sont plus nombreux dans le cerveau des jeunes patientes touchées par le syndrome de Rett. Cette substance peut provoquer des interférences entre les cellules nerveuses du cerveau, contribuant en partie aux convulsions, troubles du comportement et difficultés d'apprentissage. Le dextrométhorphane est en cours de test pour contrer ou bloquer les effets de cette substance chimique du cerveau. Le dextrométhorphane est utilisé comme médicament contre la toux[32].
La kétamine a également été testée chez un modèle animal avec des résultats prometteurs[33]. Il est rapporté que l'utilisation de kétamine chez des souris atteintes du syndrome de Rett peut inverser les anomalies de l'activité cérébrale et améliorer la fonction neurologique de ces animaux[34]. La kétamine peut avoir de puissants effets anesthésiques ; des travaux supplémentaires sont nécessaires pour établir l'innocuité et la faisabilité d'un traitement chez les patients atteints du syndrome de Rett. Un essai clinique est programmé[35].
L'étude des effets de la désipramine a été étudiée en France[36]. Lors du 4e congrès européen sur le syndrome de Rett en 2015, il a cependant été indiqué[37] que les tests n'ont pas permis de mettre en évidence d'amélioration de la fonction respiratoire par rapport à un placebo.
Dans la famille des monoamines, la société Newron Pharmaceuticals a lancé[38] en mai 2016 un essai thérapeutique sur le Sarizotan, qui permettrait d'améliorer la fonction respiratoire et de supprimer les apnées. L'essai clinique est organisé aux États-Unis, en Inde, en Italie et au Royaume-Uni[39]. Les résultats sont attendus pour la fin de l'année 2017.
Facteurs de croissance
Les agents qui favorisent le développement du cerveau et la fonction synaptique, comme l’insuline-like growth factor 1 (IGF1), sont des molécules intéressantes[40] avec des tests en cours chez des patients[41].
Neuro-immunologie
L'utilisation d'une greffe de moelle osseuse pour remplacer les cellules défectueuses du système immunitaire dans des souris modèles du syndrome de Rett a stoppé de nombreux symptômes graves de la maladie (respiration et mouvement anormaux) et considérablement étendu la durée de vie des souris Rett[42]. On connait le cas d'une fille atteinte du syndrome de Rett, traitée pour une leucémie, qui a vu ses capacités de communication accrues après une greffe de moelle osseuse, et qui a pu converser avec sa mère pour la première fois de sa vie[43].
Stimulation cérébrale profonde
L'utilisation de cette technique invasive, réservée à certains troubles (maladie de Parkinson), est actuellement à l'étude au stade pré-clinique[44].
Thérapie génique
L'activation du gène MeCP2 par l'équipe d'Adrian Bird a permis d'améliorer l'atteinte neurologique chez un modèle animal[45].
Ces travaux démontrent que la restauration du gène MECP2, même à des stades adultes, corrige plusieurs aspects de la pathologie ; ils suggèrent que la maladie peut être traitée de façon inhérente. Cela permet d'envisager plusieurs approches thérapeutiques ciblant la maladie au niveau du gène, parmi lesquelles :
- la thérapie génique, avec des recherches en cours aux États-Unis et en Europe[46] ;
- l'activation du gène MECP2 sur le chromosome X inactif[47] ;
- les médicaments de translecture translationnelle[48].
En 2017, la société AveXis a annoncé vouloir développer un programme de thérapie génique pour le syndrome de Rett[49]. Novartis, qui a racheté AveXis en 2018, avait confirmé sa volonté de poursuivre dans cette voie[50] avant d'abandonner son programme en 2021[51].
Financement de programmes de recherche
2 fondations américaines sont les premiers contributeurs des programmes de recherche sur la maladie et les traitements :
Congrès internationaux
Congrès européens :
- 2009 : Milan (Italie)
- 2011 : Kazan (Russie)
- 2013 : Maastricht (Pays-Bas)
- 2015 : Rome (Italie)
- 2017 : Berlin[54] (Allemagne)
- 2019 : Tampere (Finlande)
Congrès mondiaux :
Soutien caritatif
En 1993, l'association internationale du syndrome de Rett reçoit 25 000 dollars de la part du groupe Pearl Jam[57].
La même année, un album de 8 titres des Allman Brothers est vendu en édition limitée (15 000 exemplaires) au bénéfice de cette association[58].
L'actrice Julia Roberts plaide en octobre 2002 devant une commission du Congrès américain en faveur d'une augmentation des crédits pour la recherche sur cette maladie[59]. Elle a auparavant participé à un documentaire pour la chaîne Discovery Health Channel[60].
Histoire
Le pédiatre Andreas Rett (1924-1997) est le premier à avoir décrit ce syndrome en 1966[61].
En octobre 1983, le neuropédiatre suédois Bengt Hagberg (sv) (1923-2015) l'observe chez trente-cinq patients de sexe féminin[62]. On donnera par la suite le nom de « Syndrome de Rett » à l'affection en question.
En juin 1992, Adrian Bird découvre la protéine MeCP2[63].
Son caractère de maladie génétique a été mis en évidence en 1999 par l'équipe du professeur Huda Zoghbi de l'Institut de recherche neurologique de Houston[64].
Notes et références
- INSERM US14 -- TOUS DROITS RESERVES, « Orphanet: Syndrome de Rett », sur www.orpha.net (consulté le )
- Friederike Ehrhart, Nasim Bahram Sangani et Leopold M. G. Curfs, « Current developments in the genetics of Rett and Rett-like syndrome », Current Opinion in Psychiatry, (ISSN 1473-6578, PMID 29206688, DOI 10.1097/YCO.0000000000000389, lire en ligne, consulté le )
- (en) Ana Westenberger, Christine Klein, Valerija Dobricic et Dusanka Savic-Pavicevic, « WDR45 mutations may cause a MECP2 mutation-negative Rett syndrome phenotype », Neurology Genetics, vol. 4, no 2, , e227 (ISSN 2376-7839, DOI 10.1212/NXG.0000000000000227, lire en ligne, consulté le )
- (en) « Atypical Rett Syndrome and Intractable Epilepsy With Novel GRIN2B Mutation », sur National Center for Biotechnology Information
- (en) Silvia Russo, Lidia Larizza, Aglaia Vignoli et Isabella Moroni, « Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes », International Journal of Molecular Sciences, vol. 20, no 15, , p. 3621 (DOI 10.3390/ijms20153621, lire en ligne, consulté le )
- (en) Simranpreet Kaur, Nicole J. Van Bergen, Kristen J. Verhey et Cameron J. Nowell, « Expansion of the phenotypic spectrum of de novo missense variants in kinesin family member 1A (KIF1A) », Human Mutation, vol. 41, no 10, , p. 1761–1774 (ISSN 1098-1004, DOI 10.1002/humu.24079, lire en ligne, consulté le )
- « LE SYNDROME DE RETT », sur www.germaco.net (consulté le ).
- Einspieler C, A.M. Kerr and H. F. Prechtl (2005) "Is the early development of girls with Rett disorder really normal?" Pediatr Res57(5 Pt 1): 696-700.
- Temudo T & al. (2007). "Stereotypies in Rett syndrome: analysis of 83 patients with and without detected MECP2 mutations." Neurology68(15): 1183-1187.
- (en) « DEFINE_ME », sur ejpn-journal.com (consulté le ).
- (en) Nelson, Charles A., « Adapting the Mullen Scales of Early Learning for a Standardized Measure of Development in Children With Rett Syndrome / Intellectual and Developmental Disabilities », Intellectual and Developmental Disabilities, vol. 55, no 6, , p. 419–431 (ISSN 1934-9491, DOI 10.1352/1934-9556-55.6.419, lire en ligne, consulté le ).
- (en) « Clinicians make mistakes about intellectual impairments – as new Rett syndrome findings show », sur The Conversation (consulté le ).
- Jacky Guy, Jian Gan, Jim Selfridge et Stuart Cobb, « Reversal of neurological defects in a mouse model of Rett syndrome », Science (New York, N.Y.), vol. 315, no 5815, , p. 1143–1147 (ISSN 1095-9203, PMID 17289941, DOI 10.1126/science.1138389, lire en ligne, consulté le ).
- « Syndrome de Rett - Thérapie Génique », sur www.germaco.net (consulté le ).
- http://www.cnrs.fr/inc/communication/direct_labos/bultel.htm Elle affecte spécifiquement les filles et c’est une des principales causes de retard mental dans le monde avec 1/10000 naissances environ
- Dana Rodriguez, « Congrès polyhandicap de 2005 », dans "Le polyhandicap au quotidien: Guide à l'usage des aides médico-psychologiques" de Catherine Derouette (consulté le )
- Rett Syndrome Diagnostic Criteria Work Group (1988) Diagnostic criteria for Rett syndrome. Ann Neurol 23:425-8
- (en) Julu PO, Engerström IW, Hansen S, Apartopoulos F, Engerström B, Pini G, Delamont RS, Smeets EE, « Cardiorespiratory challenges in Rett's syndrome » Lancet 2008;371:1981-3.
- INSERM US14 -- TOUS DROITS RESERVES, « Orphanet: Syndrome de Rett », sur www.orpha.net (consulté le )
- Meir Lotan, Joav Merrick, Isack Kandel et Mohammed Morad, « Aging in Persons with Rett Syndrome: An Updated Review », The Scientific World JOURNAL, vol. 10, , p. 778–787 (ISSN 1537-744X, DOI 10.1100/tsw.2010.79, lire en ligne, consulté le )
- (en) « Rett syndrome and aging (PDF Download Available) », sur ResearchGate (consulté le )
- « Meet Ann Charlotte… – Rett Syndrome Europe », sur www.rettsyndrome.eu (consulté le )
- Wiedemann A (2017) Étude rétrospective de l’effet clinique et de la tolérance de l’acide zolédronique dans la prise en charge de l’ostéoporose de l’enfant polyhandicapé (Doctoral dissertation, Université de Lorraine) (résumé).
- Myara, L. (2009). L'orthophonie et les troubles de la déglutition et de l'alimentation dans le syndrome de Rett (Doctoral dissertation)
- (en) Inspired by love (blog de Karly Wahlin)
- Site de l’association française du syndrome de Rett
- Schwab, D., Riou, S., Fejza, A., Vial, L., Marku, J., El Husseini, W.... & Robert, Y. (2020). Le projet GazePlay et une communauté pour les personnes en situation de polyhandicap. Bulletin de la Société informatique de France, (15), 59-72|URL=https://www.societe-informatique-de-france.fr/wp-content/uploads/2020/04/1024-numero-15_Article8.pdf
- E. Smeets R. Van de Berg M. Van den Berg Leopold M.G. Curfs Gillian S. Townend, Peter B. Mar- schik. Eye gaze technology as a form of augmentative and alternative communication for individuals with rett syndrome : Experiences of families in the netherlands. 28(1):101–112, 2016
- N. Bahi-Buisson M. Voisin Université Pierre et Marie Curie (Paris) UFR de médecine Pierre et Marie Curie S. Patachon, M. Duclos. Syndrome de rett : instauration d’une communication alternative par commande oculaire : étude de cas unique. 2015. OCLC: 927105157.
- (en-US) « DAYBUE: FDA Approved Rett Syndrome », sur Acadia Pharmaceuticals (consulté le )
- (en) Jean-Christophe Roux, Diana Zala, Nicolas Panayotis, Ana Borges-Correia, Frédéric Saudou, Laurent Villard, « Modification of Mecp2 dosage alters axonal transport through the Huntingtin/Hap1 pathway », Neurobiology of disease, vol. 45, no 2, , p. 786-795 (ISSN 1095-953X, PMID 22127389, DOI 10.1016/j.nbd.2011.11.002, lire en ligne [PDF], consulté le )
- Fiche descriptive de l'essai clinique sur
ClinicalTrials.gov - (en) Miriam Kron, C. James Howell, Ian T. Adams, Michael Ransbottom, Diana Christian, Michael Ogier, David M. Katz, « Brain Activity Mapping in Mecp2 Mutant Mice Reveals Functional Deficits in Forebrain Circuits, Including Key Nodes in the Default Mode Network, that are Reversed with Ketamine Treatment », The Journal of Neuroscience, vol. 32, no 40, , p. 13860-13872 (ISSN 0270-6474 et 1529-2401, DOI 10.1523/JNEUROSCI.2159-12.2012, lire en ligne, consulté le )
- (en) [audio] Entretien entre le Pr Katz et Monica Coenraads, directrice du Rett Syndrome Research Trust|
- (en) « An Exploratory Trial of Ketamine for the Treatment of Rett Syndrome - Full Text View - ClinicalTrials.gov », sur clinicaltrials.gov (consulté le )
- « syndrome de rett », sur www.germaco.net (consulté le )
- « 4th European Congress on Rett Syndrome – Full report | Rett Syndrome Europe », sur www.rettsyndrome.eu (consulté le )
- « Newron begins STARS study of sarizotan to treat Rett syndrome », sur Drug Development Technology (consulté le )
- (en) « Evaluation of the Efficacy, Safety, and Tolerability of Sarizotan in Rett Syndrome With Respiratory Symptoms - Full Text View - ClinicalTrials.gov », sur clinicaltrials.gov (consulté le )
- (en) « IGF1 as a Potential Treatment for Rett Syndrome: Safety Assessment in Six Rett Patients », sur hindawi.com.
- (en) « Treatment of Rett Syndrome With rhIGF-1 (Mecasermin [rDNA]Injection) », sur ClinicalTrials.gov.
- (en) Noël C. Derecki, James C. Cronk, Zhenjie Lu, Eric Xu, Stephen B. G. Abbott, Patrice G. Guyenet, Jonathan Kipnis, « Wild-type microglia arrest pathology in a mouse model of Rett syndrome », Nature, vol. 484, no 7392, , p. 105-109 (ISSN 0028-0836, PMID 22425995, DOI 10.1038/nature10907, lire en ligne, consulté le )
- « Paper describes results of BMT in models of Rett Syndrome », sur News-Medical.net, (consulté le )
- (en) Stuart R. Cobb, « Cognitive disorders: Deep brain stimulation for Rett syndrome », Nature, vol. 526, no 7573, , p. 331–332 (ISSN 0028-0836, DOI 10.1038/526331a, lire en ligne, consulté le )
- (en) Guy J, Gan J, Selfridge J, Cobb S, Bird A. « Reversal of neurological defects in a mouse model of Rett syndrome » Science 2007;315:1143-7.
- (en) « A codon-optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome », sur ResearchGate (consulté le )
- (en-US) « Muting X chromosome silencers may serve as remedy for Rett | Spectrum », Spectrum, (lire en ligne, consulté le )
- Kamal KE Gadalla, Paul D Ross, Ralph D Hector et Noha G Bahey, « Gene therapy for Rett syndrome: prospects and challenges », Future Neurology, vol. 10, no 5, , p. 467–484 (ISSN 1479-6708, DOI 10.2217/fnl.15.29, lire en ligne, consulté le )
- « Avexis va faire progresser son programme de thérapie génique initié par le RSRT », sur Rett Syndrome Research Trust (consulté le )
- (en-US) « UPDATE: AVXS-201 Gene Therapy for Rett Status | RSRT », sur Rett Syndrome Research Trust, (consulté le )
- (en) Monica CoenraadsChief Executive OfficerRett Syndrome Research Trust, « Novartis Terminates Their Rett Syndrome Gene Replacement Program », sur Rett Syndrome News (consulté le )
- (en-US) « About the Foundation - Our Impact - Rettsyndrome.org », sur www.rettsyndrome.org (consulté le )
- (en-US) « About Us - Rett Syndrome Research Trust », sur reverserett.org (consulté le )
- (en) « 5. European Rett Syndrome Congress 2017 », sur www.rett2017.berlin (consulté le )
- « VIII WORLD RETT SYNDROME CONGRESS », sur www.worldcongress2016.rettsyndrome.ru (consulté le )
- (en) « 9th Rett World Congress », sur http://rettworldcongress.org/ (consulté le )
- (en) « Pearl Jam donates to Rett syndrom » The Tuscaloosa News, publié le 22 décembre 1993, consulté le 24 septembre 2012.
- [audio] The Allman Brothers Band, IRSA International Rett Syndrome Association, 1993.
- (en) CNN Politics, Julia Roberts chokes back tears before Congress, publié le 9 mai 2002, consulté le 24 septembre 2012.
- (en) « Videos : Julia Roberts In "Silent Angels" (2000) And "His Way" (2011) - AboutJulia.Com », sur AboutJulia.Com, (consulté le ).
- (en) « Article « Syndrome de Rett » », sur Who Named It?
- (en) Bengt Hagberg, Jean Aicardi, Karin Dias et Ovidio Ramos, « A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls : Rett's syndrome : Report of 35 cases », Annals of Neurology, (lire en ligne)
- (en) Joe D. Lewis, Richard R. Meehan, William J. Henzel, Ingrid Maurer-Fogy, Peter Jeppesen, Franz Klein et Adrian Bird, « Purification, sequence, and cellular localization of a novel chromosomal protein that binds to Methylated DNA », Cell, (lire en ligne)
- (en) Ruthie E. Amir, Ignatia B. Van den Veyver, Mimi Wan et Charles Q. Tran, « Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2 », Nature Genetics, vol. 23, no 2, , p. 185–188 (ISSN 1061-4036, DOI 10.1038/13810, lire en ligne, consulté le )
Voir aussi
Articles connexes
Liens externes
- Fiche grand public d'Orphanet

- Association française du syndrome de Rett
- Le syndrome de Rett sur www.autisme.qc.ca
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:312750
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005
- (en) Practical genetics dans European Journal of Human Genetics (2006) 14, 896–903 :
- (en) International Rett Syndrome Foundation
- (en) Rett Syndrome Research Trust
- (en) Rett Syndrome Association (Royaume-Uni)
- (en) Rettgirl.org, site américain d'informations sur des produits, échange de conseils entre familles, ressources sur l'alimentation, thérapies
Bibliographie
- Bévillard A & Le Pivain C (2016) Exploration des capacités communicationnelles par commande oculaire chez quatre enfants atteintes du syndrome de Rett (Doctoral dissertation).
- Buisson N.B, Hully M & Celestin E (2019) Stéréotypies et troubles moteurs dans le syndrome de Rett.
- Bumin, G., M. Uyanik, H. Kayihan, T. Duger and M. Topcu (2002). "The effect of hand splints on stereotypic hand behavior in Rett's syndrome." Turk J Pediatr44(1): 25-29.
- Dy, M. E., J. L. Waugh, N. Sharma, H. O'Leary, K. Kapur, A. M. D'Gama, M. Sahin, D. K. Urion and W. E. Kaufmann (2017). "Defining Hand Stereotypies in Rett Syndrome: A Movement Disorders Perspective." Pediatr Neurol75: 91-95.
- FitzGerald, P. M., J. Jankovic and A. K. Percy (1990). "Rett syndrome and associated movement disorders." Mov Disord5(3): 195-202.
- FitzGerald, P. M., J. Jankovic, D. G. Glaze, R. Schultz and A. K. Percy (1990). "Extrapyramidal involvement in Rett's syndrome." Neurology40(2): 293-295.
- Hagberg, B. and M. Romell (2002). "Rett females: patterns of characteristic side-asymmetric neuroimpairments at long-term follow-up." Neuropediatrics33(6): 324-326.
- Humphreys, P. and N. Barrowman (2016). "The Incidence and Evolution of Parkinsonian Rigidity in Rett Syndrome: A Pilot Study." Can J Neurol Sci43(4): 567-573.
- Naganuma, G. M. and F. F. Billingsley (1988). "Effect of hand splints on stereotypic hand behavior of three girls with Rett syndrome." Phys Ther68(5): 664-671.* Vignoli, A., F. La Briola and M. P. Canevini (2009). "Evolution of stereotypies in adolescents and women with Rett syndrome." Mov Disord24(9): 1379-1383.
- Nomura, Y. and M. Segawa (1990). "Characteristics of motor disturbances of the Rett syndrome." Brain Dev12(1): 27-30.
- Nomura, Y. and M. Segawa (1992). "Motor symptoms of the Rett syndrome: abnormal muscle tone, posture, locomotion and stereotyped movement." Brain Dev14 Suppl: S21-28.
- Nomura, Y. (2005). "Early behavior characteristics and sleep disturbance in Rett syndrome." Brain Dev27 Suppl 1: S35-S42.
- Sigafoos J & WOODYATT G (1996) Le syndrome de Rett et ses implications pédagogiques. Revue Européenne du Handicap Mental, 3(11), 21-31.
- Temudo, T., E. Ramos, K. Dias, C. Barbot, J. P. Vieira, A. Moreira, E. Calado, I. Carrilho, G. Oliveira, A. Levy, M. Fonseca, A. Cabral, P. Cabral, J. P. Monteiro, L. Borges, R. Gomes, M. Santos, J. Sequeiros and P. Maciel (2008). "Movement disorders in Rett syndrome: an analysis of 60 patients with detected MECP2 mutation and correlation with mutation type." Mov Disord23(10): 1384-1390
- Tuten, H. and J. Miedaner (1989). "Effect of hand splints on stereotypic hand behavior of girls with Rett syndrome: a replication study." Phys Ther69(12): 1099-1103.