Syndrome de Cornelia de Lange
Le syndrome de Cornelia de Lange ou syndrome de Brachmann-de Lange est une maladie génétique associant un visage caractéristique avec synophris, un retard de croissance intra utérin avec microcéphalie, un hirsutisme, un comportement agressif et des anomalies des membres supérieurs atteignant surtout les doigts allant d'anomalies très légères à une oligodactylie (diminution du nombre de doigts ou d'orteils).
| Syndrome de Cornelia de Lange | ||
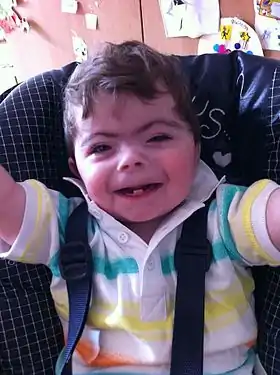 Garçon (2011) atteint du syndrome de Cornelia de Lange | ||
| Référence MIM | 122470-300590 | |
|---|---|---|
| Transmission | Dominante-Liée au chromosome X | |
| Chromosome | 5p13.1-Xp11.22-p11.21 | |
| Gène | NIPBL-SMC1L1 | |
| Mutation | Plus de 50 | |
| Mutation de novo | 99 % | |
| Anticipation | Non | |
| Porteur sain | Sans objet | |
| Prévalence | 1/10 000 à 100 000 | |
| Pénétrance | 100 % pour le NIPBL-Inconnue pour le SMC1L1 | |
| Maladie génétiquement liée | Aucune | |
| Diagnostic prénatal | Possible | |
| Liste des maladies génétiques à gène identifié | ||
Le diagnostic repose sur l'association dysmorphisme, retard de croissance, retard mental, anomalies des membres et hirsutisme. La mutation du gène NIPBL est retrouvée dans 50 % par séquençage. La mutation du gène SMC1L1 est retrouvée dans un petit nombre de cas.
Historique
Brachmann le décrit dès 1916[1] mais Cornelia de Lange en publie la description en 1933[2].
Cause
Des mutations sur plusieurs gènes ont été identifiées : sur le NIPBL, responsable de la synthèse de la delangine[3] et membre de la famille des cohésines, sur le SMC1L1[4], codant une autre cohésine, sur le SMC3 et le SMC1A[5].
Un mosaïcisme est retrouvé dans un peu moins d'un quart des cas[6].
Des mutations sur d'autres gènes donnent des syndromes apparentés, appelés « cohésinopathies », comme celles concernant le gène RAD21, entraînant une dysmorphie semblable mais moins de retard intellectuel[7].
Diagnostic
Se fait essentiellement sur la clinique :
- aspect du visage
- retard de croissance
- retard intellectuel
- anomalies des membres
- hirsutisme
L'estimation du risque peut se faire par dosage de la PAPP-A (Pregnancy-Associated Plasma Protein-A) entre la 11ème et la 14ème semaine de grossesse (la sécrétion de l'hormone protéique étant nulle donc les taux de l’hormone sont nuls).
Le retard intellectuel est constant. Les enfants atteints souffrent d'autisme ou ont des tendances auto-destructives.
Les autres anomalies trouvées dans ce syndrome comprennent des cardiopathies, des anomalies de l'appareil digestif, une surdité, une myopie, des anomalies génitales.
Il existe des formes classiques et des formes légères de ce syndrome.
Diagnostic différentiel
L'aspect de ces enfants peut se rencontrer dans :
- duplication partielle du bras long du chromosome 3
- délétion du locus q31 du chromosome 2
- syndrome de Fryns
- syndrome d'alcoolisation fœtale
Notes et références
- Brachmann W, Ein fall von symmetrischer monodaktylie durch Ulnadefekt, mit symmetrischer flughautbildung in den ellenbeugen, sowie anderen abnormitaten (zwerghaftogkeit, halsrippen, behaarung), Jarb Kinder Phys Erzie, 1916;84:225–235
- de Lange C, Sur un type nouveau de degenerescence (typus Amstelodamensis), Arch Med Enfants, 1933;36:713–719
- Krantz ID, McCallum J, DeScipio C et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B, Nat Genet, 2004;36:631–635
- Musio A, Selicorni A, Focarelli ML et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations, Nat Genet, 2006;38:528–530
- Deardorff MA, Kaura M, Yaeger D et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation, Am J Hum Genet, 2007;80:485–494
- Huisman SA, Redeker EJ, Maas SM, Mannens MM, Hennekam RC, High rate of mosaicism in individuals with Cornelia de Lange syndrome, J Med Genet, 2013;50:339–344
- Deardorff MA, Wilde JJ, Albrecht M et al. RAD21 mutations cause a human cohesinopathy, Am J Hum Genet, 2012;90:1014–1027
Voir aussi
Articles connexes
Liens externes
Bibliographie
- Robinson, L. K., Jones, K. L., Wolfsberg, E., Opitz, J. M., & Reynolds, J. F. (1985). Brachmann‐de Lange syndrome: Evidence for autosomal dominant inheritance. American journal of medical genetics, 22(1), 109-115.