Sténose et atrésie pulmonaire
Le terme sténose et atrésie pulmonaire est employé pour désigner les anomalies congénitales du cœur en rapport avec un rétrécissement de la voie d'éjection du ventricule droit du cœur. Le terme atrésie désignant une absence totale de communication entre le ventricule droit et l'artère pulmonaire.
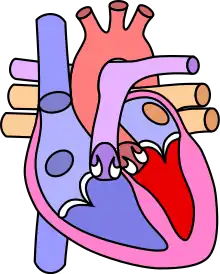
L'origine de cette pathologie est soit une fusion des trois valves de la sigmoïde pulmonaire, une hypoplasie de la voie d'éjection ventriculaire et/ou de l'artère pulmonaire ou une dysplasie de la sigmoïde pulmonaire.
Cette pathologie peut être en rapport avec une rubéole congénitale, un syndrome de Williams, un syndrome de Turner, un syndrome de Costello ou un syndrome de Noonan.
Cette sténose entraîne une hypertrophie du ventricule droit et une dilatation post-sténotique de l'artère pulmonaire qui est rarement présente avant la naissance.
Cette cardiopathie congénitale est relativement fréquente puisque celle-ci est estimée à 1 sur 1 500 naissances[1]. De nombreuses formes légères ne sont diagnostiquées que plusieurs mois après la naissance. Cette pathologie se rencontrerait chez 5 % à 10 % des enfants dans un service de cardiologie infantile
Diagnostic
Aspects échographiques avant la naissance
Le diagnostic anténatal est possible bien que plusieurs études montrent un taux de détection de cette pathologie inférieure à 10 %. Dans une étude récente norvégienne[2], le taux de détection de la sténose pulmonaire est de 25 % (1 sur 4) et de 100 % pour l'atrésie (4 sur 4) mais il s'agit d'une étude faite dans un centre hospitalier de référence. La sténose pulmonaire étant une des cardiopathies ayant un des plus faibles taux de détection avec la sténose de l'aorte et le retour veineux pulmonaire anormal total.
En fonction de la gravité, un diagnostic anténatal est possible sur les signes suivants :
- Hypertrophie du ventricule droit ;
- Hypertrophie de l'oreillette droite ;
- Diamètre de l'artère pulmonaire inférieure à celui de l'aorte ;
- L'étude du flux vasculaire en Doppler couleur constate une turbulence post-sténotique, une régurgitation au niveau de la tricuspide et souvent un flux inversé (reversed flow) du canal artériel.
Le diagnostic différentiel principal, in utero, de cette pathologie est la tétralogie de Fallot.
Gestion de la grossesse
En fonction de la sévérité de la pathologie, une insuffisance cardiaque congestive est possible avec apparition d'un hydrops fœtal et mort in utero. Bien que l'association entre une sténose pulmonaire et des anomalies chromosomiques soit rare, une étude du caryotype avec recherche de microdélétion pourra être effectuée. L'accouchement devra se faire dans un centre disposant d'un service avec cardiopédiatre.
Prise en charge du nouveau-né
Dès la naissance, l'enfant est mis sous perfusion de prostaglandine pour empêcher la fermeture du canal artériel et maintenir une perfusion pulmonaire.
Postnatal
Le tableau clinique d'un enfant porteur d'une atrésie pulmonaire est celui d'une cardiopathie cyanogène réfractaire à l'oxygénation dès les premiers instants de vie. À l'arrêt de la circulation fœtale, le canal artériel se ferme et les poumons ne reçoivent plus de sang à oxygéner, raison pour laquelle toute augmentation de la quantité d'oxygène inhalée ne traite pas l'hypoxémie. Un shunt droite-gauche s'installe si un orifice interventriculaire persiste.
L'enfant se présente cyanosé à la naissance même avec la mise sous cloche à oxygène.
Traitement
En l'absence de communication interventriculaire, une atrioseptostomie de Rashkind (septostomie atriale) est effectuée pour limiter l'insuffisance cardiaque droite et assurer le retour veineux.
Le traitement est chirurgical, et consiste :
- à ouvrir la voie pulmonaire par dilation au ballonnet (en première intention[3]) ou par abord chirurgical direct ;
- à installer un shunt de type Blalock avec la mise en place d'une prothèse systémico-pulmonaire entre une artère sous-clavière et une artère pulmonaire.
La dilatation au ballon peut être faite à tout âge, avec de bons résultats à long terme[4]. Une fuite pulmonaire peut toutefois apparaître[5].
En cas d’atrésie de la valve pulmonaire, une perforation de cette dernière, par voie endovasculaire peut être tentée, avec un risque, cependant, de plaie cardiaque[6].
Conseil génétique
En l'absence de maladie à transmission héréditaire, le risque d'avoir un autre enfant atteint de cette pathologie est de 2 % mais passe à 6 % si deux enfants sont atteints. En cas de mère enceinte porteuse de cette pathologie, le risque est de entre 4 et 6 % mais il est de 2 % si seul le père est atteint[7].
Voir aussi
Références
- Congenital Heart Disease in 56109 Births Incidence and Natural History S.C.Mitchel; S.B. Korones; H.W.rendes Circulation 1971 43: 323 - 332
- E. Tegnander, W. Williams, O.J. Johansen, H.. K. Blaas, S.H. Eik-Nes Prenatal detection of heart defects in a non-selected population of 30 149 fetuses - detection rates and outcome Ultrasound in Obstetrics and Gynecology, Volume 27, Issue 3, Pages 252-265 March 2006
- Barry OM, Bouhout I, Turner ME, Petit CJ, Kalfa DM, Transcatheter cardiac interventions in the newborn, J Am Coll Cardio, 2022;79:2270-2283
- Jarrar M, Betbout F, Farhat MB et al. Long-term invasive and noninvasive results of percutaneous balloon pulmonary valvuloplasty in children, adolescents, and adults, Am Heart J, 1999;138:950-954
- Devanagondi R, Peck D, Sagi J et al. Long-term outcomes of balloon valvuloplasty for isolated pulmonary valve stenosis, Pediatr Cardiol, 2017;38:247-254
- Petit CJ, Qureshi AM, Glatz AC et al. {https://www.ahajournals.org/doi/abs/10.1161/01.cir.89.4.1751 Technical factors are associated with complications and repeat intervention in neonates undergoing transcatheter right ventricular decompression for pulmonary atresia and intact ventricular septum: results from the congenital catheterisation research coll], Cardiol Young, 2018;28:1042-1049
- Nora JJ, Nora AH. Update on counselling the familly with a first-degree relative with a congenital heart defect Am J Med Genet 1988;29;137-142