Neurocytome
Le neurocytome (appelé aussi neurocytome central) est une tumeur cérébrale extrêmement rare diagnostiquée chez le jeune adulte (une centaine de cas rapportés dans le monde et incidence inférieure à 1/10 000 000, cette incidence est probablement bien plus importante car il s'agit des cas rapportés, l'incidence réelle est probablement entre 1/100 000 et 1/1 000 000). Sur le plan anatomique, elle est de type tissulaire, kystique et calcifiée. Sur le plan histologique, il s'agit de neurones qui se forment à partir des cellules neuronales du septum pellucidum ou de la cavité épendymaire et non pas de cellules gliales comme dans la plupart des tumeurs cérébrales[1]. La majorité des neurocytomes centraux se développent vers l'intérieur dans le système ventriculaire formant des neurocytomes intraventriculaires. Dans la majorité des cas, il s'agit d'une tumeur bénigne, mais parfois elle peut être maligne (cancéreuse).
![]() Mise en garde médicale
Mise en garde médicale
Une revue de la littérature de 2014 retrouvait 19 cas de transformation maligne décrite.
La cinétique d'évolution de cette tumeur est lente à très lente, et de plus, il semble aussi qu'elle évolue par paliers, sans que le facteur d'évolutivité ne soit connu.
De ce fait, sa classification selon l'OMS est de grade 2 (tumeur bénigne mais à faible potentiel de malignité)[2].
Cette tumeur est quasiment toujours exclusivement située dans le système ventriculaire cérébral (surtout aux niveaux des ventricules latéraux, moins souvent dans le 3e ventricule). Cette tumeur a la fâcheuse tendance à se développer au contact des foramens de Monro et ainsi à les obstruer, entraînant de ce fait une hydrocéphalie non communicante (obstructive), et engendrant cliniquement un syndrome d'hypertension intracrânienne.
La littérature médicale rapporte quelques cas de neurocytome extraventriculaire, le plus souvent malin[3].
Le traitement pour un neurocytome central implique en première intention une résection chirurgicale, avec un risque de récidive d'environ de 20% en cas d'exérèse macroscopiquement complète[4], il dépend essentiellement d'un mauvais lavage de la cavité ventriculaire en fin de procédure opératoire.
Historique
La première description de cette tumeur cérébrale remonte en 1982 par Hassoun[5]. Les neurocytomes centraux sont des tumeurs cérébrales rares qui se situent la plupart du temps dans les ventricules latéraux près des foramens de Monro. Elles ont d'abord été découvertes par Hassoun et ses collègues en 1982 et ont été classées comme tumeurs de grade II[6]. En 1985, Wilson avait également décrit un cas rare de « neuroblastome différencié » dans le ventricule latéral qui ressemble à un oligodendrogliome sur microscopie optique. Cependant, le nom neurocytome central a été donné par Hassoun[7].
Les tumeurs neuronales primitives se constituant dans le système ventriculaire ont été considérées comme rares. La plupart des cas décrits à l’époque étaient d'origine non neuronale tels que l'oligodendrogliome, l'épendymome, le méningiome, le papillome du plexus choroïde et les cellules géantes. Avec son comportement non agressif, la tumeur a souvent été appelée «neurocytome central bénin». On pense que cela se produit chez les jeunes adultes à partir des cellules neuronales du septum pellucidum et des cellules sous-dépendantes des ventricules latéraux . La plupart des incidents initiaux signalés dans le ventricule latéral étaient bénins. Cependant, au fur et à mesure que l'on recueillait plus d'informations, le nom de neurocytome central bénin a commencé à être considéré comme un nom obsolète, car ces tumeurs ne sont pas toujours bénignes ni centralisées. De nombreuses études récentes suggèrent que leur localisation, leur potentiel biologique et leur comportement clinique sont plus variables que ce qu'on pensait auparavant. Des études récentes indiquent leur emplacement inhabituel, leur comportement biologique agressif et les récidives fréquentes après la résection chirurgicale ont suscité un intérêt significatif pour diverses modalités de traitement, ainsi que sur leur terminologie, leur potentiel de lignage et leur réglementation moléculaire[7].
Incidence
Le neurocytome central représente 0,1-0,5% des tumeurs cérébrales primaires[8] - [9]. Il existe une composante génétique de la formation de ces tumeurs, ce qui entraîne une plus grande proportion de tumeurs chez les personnes de descendance asiatique que dans d'autres groupes ethniques[10]. Les neurocytomes centraux se forment principalement chez les jeunes adultes, le plus souvent pendant la deuxième ou la troisième décennie de vie[11]. Il n'y a aucune preuve que le sexe d'une personne est déterminant dans la fréquence des neurocytomes centraux[10].
Localisation
La grande majorité des neurocytomes centraux se situent entièrement dans les ventricules. Les emplacements typiques incluent quatre localisations principales :
- Dans un ventricule latéral (quel qu'il soit) autour du foramen de Monro (le plus fréquent): 50%
- Dans les deux ventricules latéraux et le 3e ventricule: 15%
- Dans les deux ventricules latéraux: 15%
- Exclusivement dans le 3e ventricule: 5%
En 2019, la littérature rapportait 8 cas de neurocytomes touchant les quatre ventricules de manière simultanée.
Anatomo-pathologie
À l'échelle macroscopique, le neurocytome central est de couleur grisâtre, ressemblant à la matière grise, et comporte des zones d'hémorragies. La tumeur est ovoïde, lobulée en formant une masse nodulaire généralement bien circonscrite. Lors de la dissection de la tumeur, les neurochirurgiens peuvent faire face à une modification de consistance, à la fois plus solide (calcifications), ou plus liquide (formation kystique). Les échantillons de tumeur colorés à l'aide de la tache H & E et examinés au microscope ont révélé que le neurocytome est une tumeur bien différenciée présentant des caractéristiques histologiques bénignes. La tumeur est composée de "cellules uniformes, de petite et de moyenne taille avec des noyaux arrondis, de la chromatine finement pointillée et des nucléoles discrets, avec un peu de cytoplasme". Le neurocytome est caractérisé par des pseudorosettes périvasculaires, des arrangements circulaires / floraux de cellules avec un petit vaisseau sanguin au centre et des petites cellules polygonales avec un halogénure périnucléaire clair. Le principal diagnostic différentiel histomorphologique est l'’oligodendrogliome. Alors que les cellules tumorales sont denses dans certaines zones, les zones avec des parties tumorales anucléées, moins denses ont été dispersées dans l'ensemble. Les zones antinucléées peuvent avoir une matrice fibrillaire fine, comme celle des régions de neuropil. Les vaisseaux longs, à paroi mince et à grandeur capillaire représentent la vascularisation du neurocytome, à ne pas confondre avec le réseau veineux profond cérébral constitutionnel qui est plus volumineux en taille et qui est septal. Ces vaisseaux sont disposés selon un motif de ramification linéaire, avec un aspect endocrinien. Les canaux vasculaires dilatés à paroi mince, ainsi que les foyers de calcification, ont été facilement identifiés dans de nombreux cas[12].
Symptômes
Les symptômes incluent l'apanage des signes d'une hypertension intracrânienne[12], à savoir céphalées persistantes, troubles de la vision (avec un œdème papillaire bilatéral au fond d'œil) ainsi que troubles de la mémoire, nausées, vomissements, trouble de la conscience…
D'autres symptômes comme des troubles moteurs (hémiparésie[13], troubles de la marche), cognitifs (attentionnels en plus de la mémoire), hémorragiques (épistaxis) ou psychiatriques (psychose[14]) peuvent être présents de manière inaugurale, mais sont moins inauguraux que les précédents.
Seule une imagerie cérébrale (TDM ou IRM encéphalique) permet de faire le diagnostic morphologique précis.
Lorsque le processus tumoral s'est installé sur plusieurs années, il peut ne pas y avoir de symptômes pendant longtemps, et l'état clinique du patient décompense alors brutalement vers un tableau d'hypertension intracrânienne, pouvant aller jusqu'à l'engagement cérébral et au décès.
Diagnostic
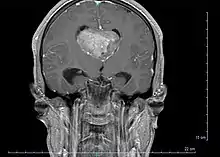
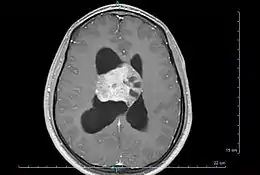
Le diagnostic est réalisé grâce à l'imagerie cérébrale, scanner ou au mieux IRM cérébrale, retrouvant cette formation hétérogène à trois composantes en général (tissulaire +/- kystique et calcique), avec rehaussement partiel à l'injection de produit de contraste.
Elle permet d'étudier les rapports avec les zones fonctionnelles du cerveau (notamment le pilier du fornix, un des composants du circuit de Papez responsable de la mémoire antérograde), le système vasculaire (réseau veineux profond), ainsi que le retentissement de la tumeur sur le cerveau (effet de masse de par l'hydrocéphalie, œdème, hémorragie, engagement…).
Seule l'anatomopathologie permet de conclure définitivement au diagnostic de neurocytome, grâce notamment à des techniques d'immunohistochimie.
Thérapie
Chirurgie
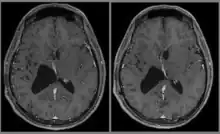
La chirurgie reste l'option de première intention[15]. L'exérèse totale doit en être l'objectif princeps.
Il existe deux voies d'abord chirurgicales :
- l'abord transventriculaire (induisant inéluctablement une épilepsie séquellaire, car la voie d'abord nécessite une corticotomie) ;
- l'abord transcalleux.
L'acte chirurgical n'est pas toujours simple. Il se fait à l'aide d'un microscope opératoire, et parfois il peut se faire à l'aide de la neuronavigation (son utilisation reste limitée car elle n'est pas aussi précise qu'elle le devrait du fait des mouvements de la tumeur baignée dans le liquide céphalo-rachidien).
L'objectif est d'enlever un maximum de tumeur sans causer de dommage aux fibres nerveuses et de respecter la vascularisation cérébrale.
On note la présence de veines profondes dans les ventricules latéraux, notamment la veine thalamo-striée, les veines choroïdiennes et la veine septale antérieure qui se réunissent pour donner la veine cérébrale interne qui draine les structures profondes du cerveau telles que le noyau caudé, le thalamus, le noyau lenticulaire, une partie du centre semi-ovale et la capsule interne. Engendrer de manière iatrogène une modification de ce drainage fait provoquer un AVC similaire à une thrombophlébite cérébrale irréversible avec la formation d'un œdème cérébral, pouvant entraîner un engorgement veineux et dans les cas les plus défavorables il peut en résulter une ischémie et un infarctus cérébral en cas d'absence de reprise d'une veine collatérale[13], aboutissant à des conséquences cliniques pouvant être parfois dramatiques (trouble de la mémoire, hémiplégie, coma, décès…).
De même, l'acte chirurgical doit respecter au maximum la structure du pilier du fornix qui tapisse la face interne du foramen de Monro sous peine de dégâts en général réversibles sur la mémoire antérograde.
Il existe aussi un risque de complication infectieuse qu'est la ventriculite bactérienne, qui se traite par une antibiothérapie adaptée et dont le retard thérapeutique doit faire craindre l'apparition de troubles cognitifs sévères (atteinte classique des ventriculites infectieuses).
Le neurocytome est en général de consistance assez molle, et donc se fragmente (par évidement central ou latéral) et se laisse aspirer assez facilement lors de l'exérèse.
On considère que c'est lors la première chirurgie où le neurochirurgien doit mettre le paquet car c'est lors de celle-ci où il a le maximum de chance de pouvoir en faire une exérèse complète, elle doit donc être la plus agressive possible dans la mesure du respect des structures constitutionnelles nobles évoqué plus haut.
L'exérèse totale reste tout le long de la prise en charge du neurocytome l'objectif tant que celle-ci semble réalisable. Souvent, les neurochirurgiens laissent un faible résidu tumoral si nécessaire, celui « collé » au tissu sain (car difficilement ou non mobilisable), et évitent ainsi au maximum les séquelles neurologiques per ou post-opératoires.
En cas d'exérèse incomplète lors de la première chirurgie, il s'avère que les reprises chirurgicales futures s'avèrent plus complexes du fait du remaniement du reliquat tumoral et de son changement fréquent de consistance, de la fibrose qui engendre des adhérences cicatricielles avec le parenchyme sain non présentes lors de la première chirurgie, et on considère en général dans ce cas qu'il est plus sage de n'intervenir que pour faire à priori une nouvelle exérèse partielle afin de ne prendre aucun risque fonctionnel neurologique pour le patient (stratégie de chirurgies itératives avec cohabitation du reliquat restant, l'indication opératoire étant alors posée lors de l'apparition de symptômes neurologique, ce qui permet d'espacer un maximum les reprises chirurgicales, le risque de transformation maligne au décours de l'évolution dans le temps étant plus que discutable et négligeable).
On note aussi que le neurocytome reste une tumeur complètement accessible chirurgicalement, et qu'en théorie, sa résection complète est réalisable.
Une rééducation (motrice et/ou cognitive) peut être nécessaire en cas de complications per ou post-opératoire.
Radiothérapie
Il n'y a pas beaucoup de preuves confirmant que la radiothérapie est un moyen de traitement efficace. Typiquement, la radiothérapie est utilisée en postopératoire en ce qui concerne l'excision partielle ou totale de la tumeur[16]. Les caractéristiques histopathologiques du neurocytome, la différenciation neuronale, la faible activité mitotique, l'absence de prolifération vasculaire de l'endothélium et la nécrose tumorale suggèrent que la tumeur peut résister aux rayonnements ionisants. Cependant, lorsque la radiothérapie conventionnelle est utilisée, ce traitement irradie le cerveau dans sa globalité et essaye d’envoyer un maximum d’énergie sur la lésion. Cette méthode utilise un programme de fractionnement standard et une dose tumorale totale de 50-55 Gy[13].
Le gamma knife, appelé aussi radiochirurgie, est une radiothérapie stéréotaxique. Elle utilise des faisceaux de rayons gamma pour délivrer une certaine dose de rayonnement à la tumeur. Le gamma knife est très efficace pour traiter le neurocytome et maintenir le contrôle de la tumeur après la procédure lorsqu'une excision complète a été effectuée. Certaines études ont révélé que le taux de réussite du contrôle des tumeurs était d'environ 90% après les cinq premières années et de 80% après les dix premières années[4] - [17]. Le gamma knife est la forme de radiothérapie la plus répertoriée pour traiter les reliquats de neurocytome après une intervention chirurgicale dans la mesure du respect des zones fonctionnelles et de la vascularisation voisine (sa marge d’erreur est proche du millimètre)[17].
Il existe aussi une autre forme de radiochirurgie stéréotaxique avec le Cyberknife, accélérateur à particules de protons, pour lequel le niveau de preuve pour le neurocytome est inconnu du fait de l'absence de publication d'études ou de cas rapportés, mais dont le rationnel intellectuel semble le placer au moins aussi bien que le Gamma Knife.
Chimiothérapie
La chimiothérapie est généralement limitée aux patients atteints de neurocytome central malin. Actuellement, les chimiothérapie utilisée contre le neurocytome comporte des molécules à base de platine, et un inhibiteur de la topo isomérase II. Les deux principaux schémas sont:
- Carboplatine + VP-16 + ifosfamide
- Cisplatine + VP-16 + cyclophosphamide
Étant donné que la chimiothérapie est utilisée dans de rares cas, il existe encore des informations à recueillir quant à l'efficacité de la chimiothérapie pour traiter le neurocytome. Par conséquent, ces recommandations doivent être considérées comme limitées et préliminaires[13].
Évolution et risque de récidive
Lorsque la tumeur se situe dans les ventricules latéraux, elle peut engendrer les difficultés suivantes : la plus contraignante se situe aux carrefours et aux cornes frontales des ventricules gauche et droit (intraventriculaire), empêchant l'évacuation du liquide céphalo-rachidien et provoquant une hypertension intracrânienne par hydrocéphalie obstructive. Son traitement quelle que soit sa modalité, peut s'accompagner de complication post-opératoire notable (œdème cérébral)
Dans les cas les plus défavorables, et selon la localisation du neurocytome, les séquelles sont accompagnées d'épilepsie chronique, d'aphasie (perte du langage), d'hémiplégie récupérable selon les cas et d'amnésie plus ou moins étendue qui occasionne parfois un état dépressif chronique avec tendances suicidaires (le malade, conscient de ses facultés diminuées, n'accepte pas cette situation irréversible).
Dans tous les cas, un examen d'IRM (bi-)annuel sera prescrit à vie, afin de surveiller le résidu tumoral (rares sont les cas où la chirurgie a permis d'enlever la totalité de la tumeur). Les céphalées peuvent perdurer pendant des années, dues au traumatisme cicatriciel post-opératoire.
S'il persiste une hydrocéphalie obstructive au décours des traitements (reliquat tumoral obstruant le foramen de Monroe), ou communicante (fibrose du foramen de Monro sur hémorragie ventriculaire ou remaniement tissulaire post-opératoire), il peut être nécessaire d'engager une nouvelle intervention chirurgicale afin de mettre en place un dispositif de drainage ventriculaire évacuant la surpression du liquide céphalo-rachidien vers la veine cave (dérivation ventriculo-atriale ou DVA) ou le péritoine (dérivation ventriculo-péritonéale ou DVP). On peut procéder dans certains cas à une fenestration du septum pellucidum, permettant de créer une dérivation ventriculaire interne entre les deux ventricules latéraux par déhisence sous réserve d'une parfaite circulation du LCR du foramen de Monro controlatéral. Il est aussi possible de procéder à une ventriculocisternostomie en cas d'obstruction de l’aqueduc de Sylvius.
On retiendra que le pronostic post-opératoire de cette tumeur est très bon en cas d'exérèse macroscopique complète.
De même, le pronostic post-opératoire en cas d'exérèse incomplète est généralement bon car la repousse de la tumeur est très lente ("inactivité" possible parfois pendant 15-20 ans).
En cas d'évolution rapide en taille de la tumeur ou de récidive clinique précoce liée à un indice de prolifération tumorale élevé, le pronostic à long terme est moins bon, et il faut discuter d'une stratégie thérapeutique plus agressive que prévue initialement associant une prise en charge plurimodale (reprise chirurgicale précoce associée à une radiothérapie stéréotaxique).
La probabilité de récidive est d'environ 20 % en cas d'exérèse complète macroscopiquement (persistance de cellules tumorales le long de l'épendyme, ce qui nécessite un bon lavage des cavités ventriculaires en fin de chirurgie pour éviter la récidive tumorale et ce, malgré une exérèse macroscopiquement complète pendant la chirurgie)[4]. Les facteurs identifiés à ce jour qui prédisent la récidive tumorale et les décès dus à une progression de cette pathologie sont un indice de prolifération tumoral élevée (Ki67 > 2 % sur la pièce opératoire), une récidive précoce de la maladie et une maladie disséminée (métastase) avec ou sans propagation de la maladie dans le liquide céphalo-rachidien (présence de cellules anormales tumorales dans le LCR à la ponction lombaire)[13].
Le risque de transformation maligne est directement corrélé à un indice de prolifération tumorale très élevé (Ki67>10-15%).
Diagnostic différentiel
- L'oligodendrocytome
- L'épendymome
- Le gangliogliome
- Les PNET
Notes et références
- Kerkeni, A.; Ben Lakhdher, Z.; Rkhami, M.; Sebai, R.; Belguith, L.; Khaldi, M.; Ben Hamouda, M. (Oct 2010). "[Central neurocytoma: Study of 32 cases and review of the literature].". Neurochirurgie. 56 (5): 408–14. . doi:10.1016/j.neuchi.2010.07.001.
- Louis David N.; Ohgaki Hiroko; Wiestler Otmar D.; Cavenee Webster K.; Burger Peter C.; Jouvet Anne; Scheithauer Bernd W.; Kleihues Paul (2007). "The 2007 WHO Classification of Tumours of the Central Nervous System". Acta Neuropathol. 114 (2): 97–109. PMC 1929165 Freely accessible. . doi:10.1007/s00401-007-0243-4.
- Agrawal P, Gupta K, Sodhi HB. Extraventricular neurocytoma: An uncommon tumor in a young boy. A review of literature. Neurol India [serial online] 2017 [cited 2017 Jun 22];65:202-5.
- Kim JW, Kim DG, Chung HT, Choi SH, Han JH, Park CK, Kim CY, Paek SH, Jung HW. Radiosurgery for central neurocytoma: long-term outcome and failure pattern. J Neurooncol (2013) 115:505-511. doi:10.1007/s11060-013-1253-9
- Hassoun, J.; Gambarelli, D.; Grisoli, F.; Pellet, W.; Salamon, G.; Pellissier, JF.; Toga, M. (1982). "Central neurocytoma. An electron-microscopic study of two cases.". Acta Neuropathol. 56 (2): 151–6. . doi:10.1007/bf00690587.
- Qian H.; Lin S.; Zhang M.; Cao Y. (2012). "Surgical management of intraventricular central neurocytoma: 92 cases". Acta Neurochirurgica. 154 (11): 1951–60. doi:10.1007/s00701-012-1446-6.
- Choudhari K. A.; Kaliaperumal C.; Jain A.; Sarkar C.; Soo M.; Rades D.; Singh J. (2009). "Central neurocytoma: A multi-disciplinary review". British Journal of Neurosurgery. 23 (6): 585–595. doi:10.3109/02688690903254350.
- Chuang, MT.; Lin, WC.; Tsai, HY.; Liu, GC.; Hu, SW.; Chiang, IC. (2005). "3-T proton magnetic resonance spectroscopy of central neurocytoma: 3 case reports and review of the literature.". J Comput Assist Tomogr. 29 (5): 683–8. .
- Garcia RM.; Ivan ME.; Oh T.; Barani I.; Parsa AT. (2014). "Intraventricular neurocytomas: A systematic review of stereotactic radiosurgery and fractionated conventional radiotherapy for residual or recurrent tumors". Clinical Neurology and Neurosurgery. 117: 55–64. doi:10.1016/j.clineuro.2013.11.028.
- Sharma, Mehar Chand; Deb, Prabal; Sharma, Suash; Sarkar, Chitra (August 2006). "Neurocytoma: a comprehensive review". Neurosurgical Review. 29 (4): 270–285. . doi:10.1007/s10143-006-0030-z.
- Hassoun J, Soylemezoglu F, Gambarelli D, Figarella-Branger D, von Ammon K, Kleihues P (1993). "Central neurocytoma: a synopsis of clinical and histological features". Brain Pathology. 3: 297–306. doi:10.1111/j.1750-3639.1993.tb00756.x.
- Li Y, Ye XF, Qian G, Yin Y, Pan QG (2012). "Pathologic features and clinical outcome of central neurocytoma: analysis of 15 cases". Chin J Cancer Res. 24 (4): 284–290. doi:10.3978/j.issn.1000-9604.2012.08.02
- Chamberlain, Marc C. Treatment of Central Neurocytoma. USC/Norris Cancer Center. Seattle Cancer Care Alliance. Feb. 20 2014.
- Ouma, JR. "Psychotic manifestations in brain tumour patients: 2 case reports from South Africa". Afr Health Sci. 4 (3): 190–194. Retrieved 5 May 2017.
- (en) Meic H Schmidt, Oren N Gottfried, Cornelia S von Koch, Susan M Chang, Michael W McDermott, « Central neurocytoma: a review », J Neurooncol, vol. 66, no 3, , p. 377-84. (PMID 15015671, DOI 10.1023/b:neon.0000014541.87329.3b)
- Haihui Chen, Rongrong Zhou, Jiayi Liu, Jintian Tang, Central neurocytoma, Journal of Clinical Neuroscience Volume 19, Issue 6, June 2012, Pages 849-853 doi 10.1016/j.jocn.2011.06.038.
- Karlsson Bengt; Guo Wan-Yuo; Kejia Teo; Dinesh Nivedh; Pan David Hung-Chi; Jokura Hidefumi; Kawagishi Jun; van Eck Albertus; Horstmann Gerhard A.; Yeo Tseng Tsai; Yamamoto Masaaki (2012). "Gamma Knife surgery for central neurocytomas". J Neurosurg (Suppl). 117: 96–101.