La microscopie en fluorescence (ou en épifluorescence) est une technique utilisant un microscope optique en tirant profit du phénomène de fluorescence et de phosphorescence, au lieu de, ou en plus de l'observation classique par réflexion ou absorption de la lumière visible naturelle ou artificielle [1],[2].
On peut ainsi observer divers objets, substances (organiques ou inorganiques) ou échantillons d'organismes morts ou vivants.



Elle fait désormais partie des méthodes de recherche classiques et de la biologie et continue à se développer avec l'imagerie moléculaire.
Sommaire
Principes
La fluorescence est la propriété que possèdent certains corps d'émettre de la lumière après avoir absorbé des photons de plus haute énergie. La microscopie en fluorescence repose sur la formation d'une image par détection de cette lumière émise. Le déplacement de Stokes décrit la différence entre la longueur d'onde absorbée par l'objet (émise par la source lumineuse du microscope) et émise par l'objet. Plus la différence entre les deux longueurs d'onde est grande plus il est facile d'observer la fluorescence.
Fluorescences primaire et secondaire
En fluorescence on distingue deux types d'objets :
- les premiers émettent de la lumière fluorescente par eux-mêmes [3], on parle de « fluorescence primaire » ou autofluorescence (chlorophylle, huile...),
- les autres doivent être combinés à une substance fluorescente pour émettre de la fluorescence on parle donc de « fluorescence secondaire » [3].
En microscopie de fluorescence, on peut donc visualiser directement des substances fluorescentes. Pour des substances, des cellules, des molécules non fluorescentes, il est nécessaire de les marquer par des substances appelées fluorochromes, comme le DAPI qui marque l'ADN et fluoresce en bleu.
Certains marqueurs génétiques comme la protéine fluorescente verte, (en anglais Green Fluorescent Protein ou GFP) sont aussi très utilisés en biologie dans des organismes génétiquement modifiés pour en produire de manière endogène. Dans ce cas, le fluorochrome est une protéine produite directement par la cellule elle-même et ne nécessite pas l'ajout de substrat. La fluorescence peut alors être visualisée directement dans les cellules vivantes, c'est un des domaines développés par l'imagerie moléculaire.
Les techniques de marquage
De nombreuses techniques de marquage peuvent être utilisées :
- Le marquage simple se fait par affinité entre un fluorochrome et la molécule à marquer.
- L'immunomarquage direct ou indirect fait intervenir un anticorps marqué.
- La technique du FISH sert à marquer des séquences nucléotidiques.
- L'utilisation de protéines de fusion (typiquement, la GFP) consiste à introduire dans la cellule à observer un gène de protéine recombinante fluorescente (par transfection ou infection), la protéine synthétisée est alors fluorescente.
- Le FLIP et le FRAP consistent à irradier une zone dont la fluorescence va disparaître. Ces techniques permettent d'étudier la diffusion des molécules marquées, en effet si des molécules se déplacent la fluorescence se répartira.
- Le FRET utilise deux fluorochromes, un donneur qui va transmettre son énergie à un autre fluorochrome accepteur. Elle permet d'étudier des interactions entre deux molécules.
- le BRET (Bioluminescence Resonance Energy Transfer), comme le FRET sinon que le donneur est bioluminescent (luciférase).
Excitation monophotonique
On peut exciter les substances fluorescentes par une excitation monophotonique. On utilise pour cela une lumière d'excitation dont la longueur d'onde excite directement le fluorophore. Donc, la fluorescence émise peut provenir de toute l'épaisseur de l'échantillon traversée par le faisceau d'excitation. L'élément clé de ce microscope confocal est alors représenté par une "fenêtre" (un sténopé ou un iris confocal) placée devant le détecteur qui élimine la fluorescence provenant des régions non focales. L'observation de signaux de fluorescences repose sur cinq éléments :
- une source de lumière pour l'excitation,
- un fluorophore,
- des filtres pour séparer les photons d'émission des photons d'excitation,
- un sténopé,
- un détecteur pour transformer le signal lumineux des photons en signal électrique.
Excitation multiphotonique
Cette excitation consiste en l'absorption quasi simultanée de plusieurs photons d'excitation d'une longueur d'onde proche d'un multiple de l'excitation optimale à un photon. On utilise pour cela un laser pulsé dans des fréquences proches de l'infrarouge. Dans ce cas, seul le point de focalisation du faisceau laser est excitateur (densité de photon suffisante pour coupler l'énergie d'excitation). Bien souvent les applications sont limitées à la microscopie biphotonique (excitation du fluorophore par deux photons).
Ce système est considéré comme une évolution technologique importante pour trois raisons majeures :
- Il n'existe pas d'iris confocal (sténopé) dans un microscope biphotonique. On ne peut donc plus parler de microscopie confocale, bien qu’on emploie quelquefois le terme de microscopie confocale multiphotonique de manière abusive.
- L'utilisation d'une lumière d'excitation à une longueur d'onde élevée (> 900 nm) assure une plus grande pénétration à l'intérieur de l'échantillon (jusqu'à 500 µm au lieu de 150 µm) offrant la possibilité de travailler sur des échantillons plus épais.
- L'excitation des fluorophores étant limitée au point de focalisation du faisceau laser, le risque de photoblanchiement est réduit.
En pratique, le rendement d'émission de fluorescence est moins bon qu'un confocal simple photon et le rapport signal/bruit est plus faible. Ainsi, il ne montre que peu d'avantage pour l'observation de cellules en culture ou de coupes de tissu (50-70 µm d'épaisseur).
Cette technique est naturellement utilisée pour l'excitation simultanée de plusieurs fluorophores à spectres d'émission différents.
Une variante de cette technique est la microscopie à fluorescence par excitation multiphotonique multifocale. Le principe est identique, mais le faisceau laser est divisé en plusieurs faisceaux ce qui permet de balayer simultanément plusieurs points. Ceci permet de diminuer le temps d’acquisition des images.
Différents types de microscopes
Les techniques de fluorescence peuvent être utilisées avec différents types de microscope :
- un microscope optique classique. Il est également courant de faire passer la lumière excitatrice par l'objectif et non pas par-dessous le spécimen. On parle alors de microscopie à épifluorescence.
- un microscope confocal à balayage laser. Cette association est la plus courante. Le microscope confocal atteint une résolution bien meilleure que le microscope optique classique, et permet de réaliser des images en trois dimensions de l'objet.
- un microscope de fluorescence par réflexion totale interne. Il permet notamment d'obtenir une meilleure profondeur de champ (200 nm) que la microscopie confocale (600 nm), mais uniquement à la base de l’échantillon, et plus précisément au niveau de l’interface entre l'échantillon et le support.
- la spectroscopie de corrélation de fluorescence (FCS) sert à étudier la diffusion de molécules et les interactions moléculaires. Elle est souvent associée à la microscopie confocale à balayage laser. [1]
Le filtrage dans un microscope à épifluorescence [1]

Ces appareils possèdent des pièces interchangeables en forme de cube disposées sur une tourelle rotative ou sur une tirette selon les fabricants.
Ces « cubes » filtrent la lumière allant vers l'objectif et allant vers l'observateur ou le capteur, selon les fluorescences recherchées.
Ils comportent 2 filtres et un miroir particulier, « dichroïque » qui réfléchit certaines longueurs d'onde et qui est traversé par d'autres.

Vers la source se trouve le filtre d'excitation.
Côté observateur se trouve le filtre barrière.
Les cubes sont répertoriés selon les lumières d'excitation: ultraviolet, violet, bleu, vert[4]...
On utilise cette technique, notamment pour observer des monocouches lipidiques auxquelles on a ajouté une sonde lipidique fluorescente. On observe une fluorescence selon la phase dans laquelle se trouve le lipide principal. En général, la sonde est soluble dans la phase liquide-expansé (LE) mais pas dans les phases gazeuse (G) ou solide (S). Ceci permet d'observer les macrostructures formées par la monocouche. Ci-contre, on observe une monocouche lipidique à l'équilibre par microscopie à épifluorescence. La sonde fluorescente lipidique est soluble dans la phase LE mais pas dans la phase G. Le résultat est qu'on observe, à l'équilibre, des sortes de bulles (mais une « bulle » est un concept tridimensionnel) dont les parois sont constituées du lipide en phase LE et l'intérieur en phase G. Si l'on prend un exemple tridimensionnel, cela reviendrait à prendre de la mousse de savon et à en couper une très fine tranche.
Limites de la microscopie en fluorescence
- Comme toute technique de microscopie optique classique, la microscopie en fluorescence est limitée par la diffraction de la lumière. Son pouvoir de résolution est donc de 200 nm environ (voir microscope optique).
Cette limite de résolution restreint l'utilisation de la microscopie en fluorescence pour l'étude des interactions protéines-protéines. En effet, les protéines ont une taille caractéristique de 0,1 à 1 nm, on ne peut donc pas montrer par cette technique qu'il y a contact entre deux protéines. C'est pourquoi, pour étudier l'interaction protéine-protéine, il faut s'en remettre à d'autres techniques exploitant le phénomène de la fluorescence (FRET) ou bioluminescence (BRET). - les colorants organiques visibles dans le proche infrarouge (les plus utiles pour observer à travers les tissus) sont peu nombreux et assez faiblement fluorescents.
- les colorants organiques perdent leur fluorescence en vieillissant (phénomène dit de « photoblanchiment »).
- les colorants sont observés avec des filtres et ont des propriétés spectrales telles qu'il est difficile d'observer simultanément plusieurs couleurs ; et leurs maxima d'émission sont spectralement proche de leurs maxima d'absorption.
- les nanocristaux de semi-conducteurs sont très fluorescents, et feraient sans doute des sondes très performantes, mais ils sont probablement « nanotoxiques ».
Galerie illustrative

Imagerie en épifluorescence de trois composantes d'un cellule cancéreuse humaine en cours de division. L'ADN apparaît bleu, une protéine dite INCENP apparaît en vert, et les microtubules en rouge. Chaque fluorophore a été imagé séparément, avec une longueur d'onde d'excitation spécifique et des filtres, puis une image a été recomposée, à partir des photos prises par la caméra CCD.
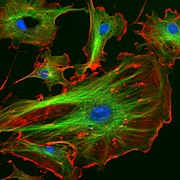
Cellules endothéliales d'une artère pulmonaire bovine (noyaux colorés en bleu avec du DAPI, microtubules marqués en vert par un anticorps lié au FITC et filaments d'actine marqués en rouge par de la phalloïdine liée à la protéine TRITC).
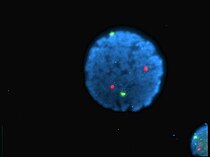
Noyau de lymphocyte humain coloré au DAPI avec les chromosome 13 (vert) et 21 (rouge) par des sondes hybridées aux centromères (hybridation in situ en fluorescence).

Membrane cellulaire de levure, mise en évidence de structure (en jaune) obtenue par fusion de protéines de la membrane avec deux marqueurs fluorescents (RFP et GFP).

Détection de la molécule YFP dans une cellule cancéreuse humaine, à une échelle nanométrique.

Noyau d'une cellule de cancer des os (technique dite dual-color localization microscopy ou 2CLM pour les anglophones). Image obtenue par fluorescence de marqueurs fusionnés avec les protéines GFP et RFP, pour 120 000 molécules localisées dans une zone restreinte (de 470 µm 2).






Bibliographie
Notes et références
- Spring KR, Davidson MW ; Introduction to Fluorescence Microscopy ; Nikon Microscopy (consulté le 28/09/2008).
- The Fluorescence Microscope , The Nobel Foundation ; Microscopes—Help Scientists Explore Hidden Worlds (consulté le 28/09/2008).
- Voir exemples dans la Galerie des images de microscopie à fluorescence primées par Nikon.
- Scordato A., Schwartz S.; Fluorescence filter combinations ; Nikon Microscopy (consulté le 09/11/2010).