Incontinentia pigmenti
L'incontinentia pigmenti (IP) ou syndrome de Bloch-Sulzberger, est une maladie génétique rare dite « orpheline » de type héréditaire, qui affecte principalement la peau, les dents, les yeux et le système nerveux central. Son nom provient des anomalies observées dans la peau à l’examen microscopique (incontinence pigmentaire).
| Incontinentia pigmenti | ||
 Incontinentia pigmenti sous l'aisselle d'une fillette de trois ans. Les « taches » adoptent les lignes de Blaschko[1]. | ||
| Référence MIM | 308300 | |
|---|---|---|
| Transmission | Génétiquement | |
| Chromosome | X | |
| Nombre de cas | 700 | |
| Liste des maladies génétiques à gène identifié | ||
| Spécialité | Génétique médicale |
|---|
| CISP-2 | S83 |
|---|---|
| CIM-10 | Q82.3 |
| CIM-9 | 757.33 |
| OMIM | 308300 |
| DiseasesDB | 29600 |
| MedlinePlus | 001583 |
| eMedicine |
1176285 article/1176285 |
| MeSH | D007184 |
| GeneReviews |
![]() Mise en garde médicale
Mise en garde médicale
Elle ne doit pas être confondue avec l'hypomélanose de Ito (HI), anciennement dénommée incontinentia pigmenti achromians dont les symptômes sont proches.
L'incontinentia pigmenti est causée par une déficience du gène nemo codant la protéine NEMO (pour NF-κB Essential Modulator) qui est une sous-unité régulatrice d’une protéine kinase (IKK) impliquée dans la voie de signalisation des facteurs de transcription ; cette protéine contrôle donc normalement la multiplication cellulaire et la réponse immunitaire et inflammatoire en limitant la mortalité cellulaire (apoptose) dans les tissus concernés ; cependant son absence entraîne aussi la dégénérescence tissulaire par apoptose[2].
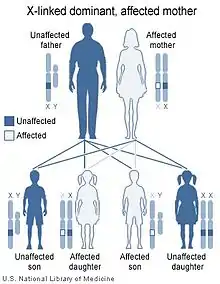
La maladie peut apparaître par mutation du gène nemo, chez les filles, et rarement chez les garçons.
Le gène nemo est dominant et lié au chromosome X. La maladie n'est héritée que par les filles car les fœtus mâles en ayant hérité ne survivent pas.
Historique
La maladie a d'abord été signalée par le dermatologue Suisse Bruno Bloch en 1926 puis par le dermatologue américain Marion Suzberger en 1928[3]. Le gène nemo a été découvert en 1998.
Épidémiologie

Une personne sur 143 000 est touchée[4]. Plus de 50 % des cas relevés sont dus à des mutations apparues de novo[4]. Globalement (hérédité + cas de novo), incontinentia pigmenti touche surtout les filles (95 % des cas) mais peut aussi atteindre les garçons[5].
Dans 5 % des cas, le fœtus mâle peut être atteint par mutation (plus ou moins de tissus sont touchés selon le stade de développement auquel a lieu la mutation) et est éliminé naturellement. Il existe cependant des exceptions correspondant à des caryotypes « 47, XXY » ou Syndrome de Klinefelter où la grossesse est menée à son terme[6] ; ces garçons (individus « mosaïque ») ne transmettent pas la maladie à leurs garçons mais peuvent la transmettre ou non à leurs filles selon que leurs tissus reproducteurs sont touchés ou non. Pour le savoir, il faut procéder à une analyse génétique des spermatozoïdes. Les symptomes éprouvés par les garçons mutants diffèrent sensiblement de ceux des filles d'où une reconnaissance tardive de la prévalence de la maladie chez les garçons.
Les filles de mère porteuse ont une chance sur deux d'être touchéees par la maladie ; les fœtus mâles touchés (50 % des garçons) sont non viables et sont éliminés naturellement. Pour déceler la maladie chez ces filles, un prélèvement peut être pratiqué in utero, afin de déterminer si le fœtus a hérité du gène ou pas[4], à trois mois (diagnostic prénatal) .
Physiopathologie
Incontinentia pigmenti est causée par une mutation sur le gène NEMO (Xq28). Ce gène est situé sur le chromosome X, la transmission de cette maladie est dominante et dite "liée à l'X". La mutation dont l'ampleur est variable (le gène est plus ou moins abîmé) affecte la production de la protéine Nemo impliquée comme régulateur du système NF-κB dont le fonctionnement précis est mal connu[2]. De plus, le phénomène d'inactivation du chromosome X ou lyonisation chez les femelles fait que les tissus sont touchés de manière aléatoire (atteintes en mosaïque). Le diagnostic prénatal permet de déceler la maladie mais en aucun cas d'en prévoir la gravité qui est très variable.
La maladie entraîne la dégénérescence de tissus comme la peau, et les vaisseaux sanguins du fond de l'œil et parfois du cerveau, dans ce cas avec des conséquences potentiellement graves (convulsions, handicap mental, ...)[6].
Elle est la cause d'anomalie du système pilaire (plages glabres) et des ongles.
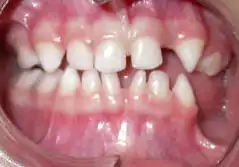
La peau s'autorépare en grande partie par élimination des cellules touchées au bout de quelques mois[6] si le sujet est maintenu dans de bonnes conditons d'hygiène (voir Examen clinique). La formule dentaire est souvent modifiée : certaines dents manquent et d'autres ont une forme conique (voir photo). Les dégâts entraînés sur la dentition peuvent être atténués par l'orthodontie[1]. Ceux entraînés sur les yeux et le cerveau sont irréversibles. Un suivi ophtalmologique est nécessaire, il peut permettre d'éviter des aggravations comme un décollement de rétine.
À noter que 41 % des patients n'ont aucune atteinte rétinienne, 7 % ont de la forte myopie et 43 % un strabisme.
Diagnostic
Outre le diagnostic prénatal, des diagnostics préimplantatoires sont envisagés dans certains pays. Ces diagnostics reposent sur l'identification du gène NEMO. Le diagnostic après naissance reposent sur l'examen clinique et doit être effectué dès la naissance, si possible
Examen clinique
L'examen de la peau à la naissance est essentiel car des lésions sont présentes à la naissance ou dans les 15 premiers jours chez 90 % des filles touchées[6].
Les lésions cutanées se présentent à quatre stades[6] :
- stade vésiculobulleux : placards (étendues plus ou moins larges de peau) rouges légèrement surélevés qui démangent et qui se recouvrent de vésicules et de bulles (cloques) bien souvent présents dès la naissance ;
- stade papuloverruqueux : apparition de lésions de type verruqueux (apparition de verrues plus ou moins importantes) et de lichen sur les mains et les pieds à l'âge de deux à cinq mois ; cette attaque cesse à la fin des premiers mois et ne laisse le plus souvent que peu de séquelles
- stade pigmenté : cette pigmentation prend un aspect en éclaboussures ou en jets d’eau. Elle peut être constatée à la naissance chez 5 à 10 % des patientes, mais ne survient le plus souvent qu’entre six et douze mois de vie ;
- stade atrophique : l’atrophie (amincissement cutané) survient avant la régression de l’hyperpigmentation. Elle prend l’aspect chez l’adolescente et l’adulte de bandes plus claires et dépilées. Elles sont plus visibles sur le cuir chevelu ou derrière les mollets. Généralement, à l’âge adulte, l’atteinte cutanée à presque complètement disparu et peut même passer totalement inaperçue si elle n’est pas soigneusement recherchée.
On observe également des manifestations extracutanées, surtout ophtalmologiques (anomalies vasculaires rétiniennes, décollement de rétine, etc.) dentaires (anodontie partielle, dents coniques, alignement anormal), sur les ongles et neurologiques (retard mental, convulsions, etc.).
L’examen cutané permet le diagnostic. On constate l’apparition précoce de vésicules et de pustules comparables à celle de la varicelle, sur la peau du nouveau-né de sexe féminin.
Examens complémentaires[6]
Les examens complémentaires incluent un suivi en ophtalmologie afin d'apprécier l'atteinte rétinienne, le cas échéant. Il est recommandé de faire un suivi : aussitôt après la naissance, tous les mois jusqu'à quatre mois, tous les trois mois jusqu'à un an et deux fois par an jusqu’à l’âge de trois ans.
Différents changements rétiniens, souvent asymétriques, peuvent être vus. Ils commencent dès les premières semaines après la naissance et progressent au cours des mois :
- grade 1 : changement de l'épithélium pigmentaire, pigmenté ou non avec des marbrures ;
- grade 2 : vascularisation ou non d'une crête temporale, avec ou sans traction rétinienne ou du vitrée, une gliose ou fibrose, parfois avasculaire dans la périphérie de la crête ;
- grade 3 : décollement de la rétine et membrane rétrocristalline.
Prise en charge
Aucun traitement médical n'est disponible[2]. Suivant les effets sur le corps du patient (dents, œil, etc.) il n'existe que les traitements locaux (orthodontie, correction laser, etc.) ; concernant les problèmes de peau tels que les plages d'alopécie ou traces d'« éclaboussures » sur les mollets, aucun traitement n'existe pour le moment.
Associations concernant l'IP
L'organisation IPIF (International Incontinentia Pigmenti Foundation) a été fondée en 1995 ; le consortium IP permet des échanges entre scientifiques ou médecins de France, Italie, Royaume-Uni, Allemagne, États-Unis[7].
Une association française « Incontinentia Pigmenti France » propose aux malades et aux familles de rompre l'isolement, se regrouper et diffuser de l'information[8]. Il n'existe pas d'association similaire en Belgique.
Notes et références
- (en) Kitakawa D., Fontes P.C., Magalhães F.A., Almeida J.D. et Cabral L.A., « Incontinentia pigmenti presenting as hypodontia in a 3-year-old girl: a case report », J. Med. Case Rep., no 3, , p. 116 (PMID 19946534, DOI 10.1186/1752-1947-3-116, lire en ligne).
- Gilles Courtois, « Les nouvelles fonctions de NEMO, la sous-unité régulatrice de la kinase activant NF-κB », sur Med.Sci (Paris), (consulté le )
- (en) « Bloch-Sulzberger pigment dermatosis », sur whonameit (consulté le )
- « Incontinentia pigmenti », sur orpha.net, (consulté le )
- (en) Gupta K.D., Padhiar B.B., Karia U.K. et Shah B.J., « Case reports of incontinentia pigmenti in males », Indian J. Dermatol., vol. 58, no 4, , p. 328 (PMID 23919034, DOI 10.4103/0019-5154.113998, lire en ligne).
- (en) Vanessa Ngan, « Incontinentia pigmenti », sur Derm.net NZ, (consulté le )
- « IPIF », sur ipif (consulté le )
- « Incontinentia pigmenti France », sur incontinentia-pigmenti.fr.
Articles connexes
Liens externes
- Incontinentia pigmenti, Orphanet