Fibrodysplasie ossifiante progressive
La fibrodysplasie ossifiante progressive (FOP), appelée également myosite ossifiante progressive, maladie de l'homme de pierre ou maladie de Münchmeyer, se caractérise par une ossification progressive des muscles squelettiques et des tendons qui les rattachant aux os, le plus souvent précédée de poussées inflammatoires. C’est une maladie génétique puisqu’elle met en cause un gène : ACVR1 ; il s’agit d’une maladie orpheline qui touche 1,3 personne par million[1].
| Spécialité | Rhumatologie |
|---|
| CISP-2 | L99 |
|---|---|
| CIM-10 | M61.1 |
| CIM-9 | 728.11 |
| OMIM | 135100 |
| DiseasesDB | 8732 |
| eMedicine | 1112501 |
| MeSH | D009221 |
![]() Mise en garde médicale
Mise en garde médicale
Épidémiologie
La fibrodysplasie ossifiante progressive est une affection extrêmement rare ; 97 % des cas rapportés dans le monde sont liés à la mutation R206H sur ACVR1/ALK2[2] - [3] - [4]. 3 % des cas sont des variantes génotypiques ou phénotypiques[4].
Définition, diagnostic
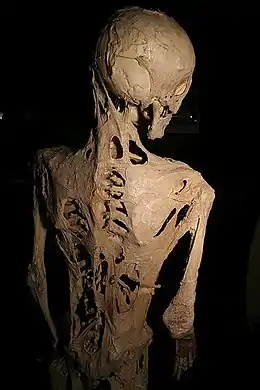
La suspicion de la FOP peut avoir lieu dès la naissance devant la malformation spécifique du gros orteil ou plus tard, dans d’autres cas, avec les gonflements tissulaires ou raideurs anormales. On assiste alors à la transformation des muscles squelettiques, des tendons et des ligaments en os hétérotopique, ce qui rend progressivement tout mouvement impossible. Les ossifications commencent vers l’âge de deux à six ans, souvent à la suite d'un traumatisme ou d'une infection. Les premières parties du corps à être ossifiées sont les muscles de la nuque puis ceux du cou ; l'ossification se poursuit progressivement le long du dos et atteint les articulations des bras et des jambes ainsi que les parties abdominales. Seuls les muscles cardiaques, la langue et le diaphragme sont toujours épargnés. L'ossification, dans la majorité des cas, semble suivre le schéma de développement squelettique embryonnaire. Mais les poussées osseuses sont imprévisibles, parfois rapprochées, parfois plus espacées.
Les symptômes tels que la malformation du gros orteil peuvent être explorés à l'aide des radiographies, mais les gonflements des tissus sont parfois pris pour des tumeurs, entraînant une mauvaise prise en charge de la maladie. La biopsie des tissus suspects entraîne à elle seule l'aggravation de l'état du patient au même titre que les chimiothérapies et les amputations inutiles. En dépit des tests génétiques aujourd’hui disponibles, le diagnostic n'est pas évident en raison de la rareté de cette affection.
La durée de vie des patients est relativement limitée ; l’espérance de vie est de 56 ans[5].
Cause
Le gène responsable de la FOP, identifié en 2006, est nommé ACVR1 (pour Activin Receptor Type 1A) : il est situé sur le bras long du chromosome 2. Ce gène code le récepteur au BMP : Bone Morphogenetic Protein, c’est une protéine qui induit la formation des os et du cartilage. Ce gène est commun à tout individu mais la maladie survient si et seulement si l’une des deux copies de ce gène contient une anomalie.
En temps normal, les cellules des muscles squelettiques et des os présentent des récepteurs ACVR1 codés par le gène ACVR1 lui-même. Dans les cas sains, on a des antagonistes aux protéines BMP qui empêchent les cellules des muscles squelettiques de se différencier en cellules cartilagineuses.

Dans le cas de la FOP, on suppose qu’il y aurait une surproduction de protéines BMP et une sous-production d’antagonistes de cette protéine. De plus, la mutation du gène ACVR1 entraînerait une augmentation du nombre de récepteurs aux BMP à la surface des cellules musculaires squelettiques. Ceci déclencherait chez ces cellules une différenciation en cellules cartilagineuses. Leur devenir est la transformation en cellules osseuses.
Le défaut génétique est une substitution de l’arginine par l’histidine. En position 206 de la protéine ACVR1, le codon CAC (responsable de la présence de l’histidine) remplace un codon CGC codant l’arginine chez le patient sain.
Il y a différents types de cas :
- cas sporadiques : chez qui la mutation survient spontanément au cours de l’embryogenèse ;
- cas familiaux : chez qui cette anomalie génétique est transmise par l’un des parents.
Prise en charge thérapeutique
Il n’existe pas de traitement curatif ; la prise en charge reste symptomatique. Les traitements pharmacologiques servent, dans un premier temps, à réduire les douleurs, avec notamment l’administration d’analgésiques, de myorelaxants, et la cryothérapie. De plus, ils servent aussi à réduire l’inflammation avec de fortes doses de corticoïdes, prescrites lors de chaque poussée de la maladie, dès les premières 24 heures (prédnisone 2 mg·kg-1) pendant quatre jours puis diminution progressive sur 10 jours.
Cependant, ces traitements ne permettent pas la guérison de la maladie. En effet, les patients ont une espérance de vie de 56 ans[5], et ceux arrivant en fin de vie sont dans un fauteuil adapté. Dans la plupart des cas, les patients meurent de complications thoraciques (insuffisance respiratoire) quand l'ossification atteint les muscles intercostaux.
La prévention des poussées de la maladie est importante. Il convient d’éviter les traumatismes des tissus mous (chute, chocs, étirements, etc.), les sports de contact, les injections intramusculaires ou sous-cutanées, les piercings, les tatouages, etc.
Des essais cliniques de plusieurs traitements potentiels sont en cours de test en 2019[6].
Recherche
Les recherches en cours permettent de retrouver l'espoir d'un traitement efficace de la FOP.
Plusieurs essais cliniques prometteurs ont été lancés depuis 2016.
Le 29 juillet 2020, une campagne de levée de fonds a été lancée par l'IFOPA, pour permettre la création d'une subvention d'une thérapie génique ciblée.
Notes et références
- « Espaces documentaires », sur BNDMR.fr,
- (en) Kaplan FS, Pignolo RJ, Shore EM, « The FOP metamorphogene encodes a novel type I receptor that dysregulates BMP signaling », Cytokine Growth Factor Rev, vol. 20, nos 5-6, , p. 399-407. (PMID 19896889, PMCID PMC3515059, DOI 10.1016/j.cytogfr.2009.10.006, lire en ligne [html])
- (en) Kaplan FS, Lounev VY, Wang H, Pignolo RJ, Shore EM, « Fibrodysplasia ossificans progressiva: a blueprint for metamorphosis », Ann N Y Acad Sci, no 1237, , p. 5-10. (PMID 22082359, PMCID PMC3502040, DOI 10.1111/j.1749-6632.2011.06195.x, lire en ligne [html])
- (en) Zhang W, Zhang K, Song L, Pang J, Ma H, Shore EM, Kaplan FS, Wang P, « The phenotype and genotype of fibrodysplasia ossificans progressiva in China: a report of 72 cases », Bone, vol. 57, no 2, , p. 386-91. (PMID 24051199, DOI 10.1016/j.bone.2013.09.002, lire en ligne [html])
- « Maladie de l'homme de pierre : symptômes, évolution, définition », sur sante.journaldesfemmes.fr (consulté le )
- Marie McCullough, Therapies in sight for FOP, a disease that turns muscle to bone, Philadelphia Inquirer, 29 February 2019.
Sources
- Centre de Référence des maladies osseuses constitutionnelles - (Hôpital Necker, Paris, France)
- FOP France
- IFOPA (États-Unis)
- Prise en charge d'un patient atteint d'une fibrodysplasie ossifiante progressive
- AFM Telethon
- Orphanet
- (en) Orphanet journal of rare diseases
- Institut de Myologie
- Tunésie Orthopédique