Chromatographie d'exclusion stérique
La chromatographie d'exclusion stérique (SEC pour size exclusion chromatography en anglais) est une méthode de chromatographie en phase liquide permettant de séparer des macromolécules en fonction de leur volume hydrodynamique. Elle est notamment utilisée pour faire l'étude de polymères. Suivant la nature des deux phases en présence, elle est encore désignée par chromatographie sur gel perméable (GPC pour gel permeation chromatography) ou par filtration sur gel (GFC pour gel filtration chromatography).
Principes de l'exclusion stérique
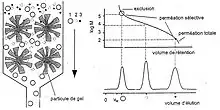
Contrairement aux méthodes de chromatographie d'affinité (comme l'high performance liquid chromatography), le principal phénomène physique permettant la séparation des différentes macromolécules constituant le polymère n'est pas basé sur l'affinité chimique avec le support, mais idéalement sur la taille des macromolécules en solution (leur volume hydrodynamique). En réalité, ces deux mécanismes de séparation sont continuellement en compétition au sein de la colonne; les conditions expérimentales sont alors établies pour minimiser les mécanismes enthalpiques d'affinité au profit de ceux entropiques d'exclusion stérique.
Le mécanisme d'exclusion stérique est fondé sur l’équilibre thermodynamique entre deux phases, le solvant interstitiel dans le volume mort et le solvant contenu dans le volume poreux . Si est le coefficient de partage, le volume d’élution d’une macromolécule est défini par :





L’évolution du coefficient de partage en fonction, d’une part de la taille de la chaîne macromoléculaire et de sa topologie, d’autre part de la distribution de la taille des pores, a une origine purement entropique. La fonction de partage d’une chaîne de taille donnée, à l’intérieur d’un pore de volume dont la paroi est impénétrable, peut être obtenue par l’énumération de toutes les conformations possibles prises au hasard. Les macromolécules dessinées en ligne continue correspondent aux conformations autorisées et les macromolécules dessinées en pointillé correspondent aux conformations impossibles car franchissant la paroi. Le coefficient de partage est lié au rapport des nombres des conformations autorisées et impossibles.
Suivant leur taille, les molécules éluées peuvent plus ou moins pénétrer dans les pores de la phase stationnaire, un gel polymère dont sont remplies les colonnes. Ainsi, les molécules les plus petites sont davantage retenues que les plus grosses, d'où un temps de rétention plus long pour les premières que pour les secondes. Chaque colonne possède un domaine de séparation spécifique: les macromolécules dont le volume hydrodynamique est trop important éluent dans le volume mort, tandis que celles trop petites éluent avec l'éluant en fin d'injection. L'analyse de données hors de ce domaine de séparation n'apporte donc aucune information.
En sortie de colonne, des détecteurs (réfractomètre, diffusion de la lumière, viscosimètre) fournissent des données relatives aux macromolécules sortant de la colonne à un instant donné.
Étalonnage de la colonne
Étalonnage conventionnel
Pour obtenir des valeurs approchées des masses molaires moyennes des polymères, il est nécessaire d'établir une ou plusieurs courbes d'étalonnage indiquant la masse molaire moyenne en fonction du volume hydrodynamique des macromolécules.
L'étalonnage est réalisé en injectant une série d'échantillons de polymères d'architecture, de faible polydispersité et de masse molaire moyenne connue. Les standards utilisés sont généralement des polystyrènes ou des polyméthyméthacrylates linéaires; le choix du standard se fait en fonction du système éluant/colonne. Le tracé du logarithme de leur masse molaire moyenne en fonction du volume de rétention (calculé à partir du temps de rétention expérimental et du débit choisi) permet d'établir la courbe d'étalonnage. Cette courbe peut être modélisé par un polynôme d'ordre 3. Le volume hydrodynamique peut être différent pour deux macromolécules de même masse molaire moyenne mais d'architecture différente. Une macromolécule non linéaire (par exemple, en peigne) occupera un plus grand volume qu'une macromolécule linéaire de même masse molaire moyenne et sera donc éluée plus rapidement même si les deux masses molaires sont identiques. Il faut donc réaliser différentes courbes d'étalonnage pour chaque type d'architecture du même polymère.
Étalonnage universel
La méthode de calibration conventionnelle n’est valable uniquement que pour un couple polymère-solvant donné, chaque couple nécessitant sa propre courbe d’étalonnage. Pour s'affranchir de ce problème, il est possible de réaliser un étalonnage universel permettant de déterminer les masses molaires moyennes de polymères d'architecture ou de composition chimique différentes.
La méthode la plus simple pour réaliser la calibration universelle est d'utiliser l'équation de Mark-Houwink-Sakurada, si les coefficients de Mark-Houwink-Sakurada sont connus pour le couple polymère/éluant/température de colonne. Cette méthode ne s'applique toutefois pas aux oligomères et aux polymères branchés dont les mécanismes de séparation dans la colonne ne peuvent uniquement être expliqués par l'exclusion stérique.
L'autre solution consiste à utiliser un viscosimètre, ce qui permet d'établir une relation directe entre le volume hydrodynamique et la masse molaire moyenne d'un polymère.
Appareillage
L’appareillage de SEC est un appareillage semblable à ceux utilisés en HPLC classique et est très largement disponible auprès de nombreux fournisseurs. La SEC est constitué d’un réservoir de solvant, d’un système de pompage, d’un injecteur, d’un jeu de colonnes et de détecteurs disposés en sortie de colonne.
Solvant
Le solvant est filtré par un filtre en ligne et ensuite dégazé à l’aide d’un dégazeur connecté en série.
Système de pompage
Le système de pompage doit être très précis et capable de travailler à haute pression (jusqu'à 200 à 300 bar). Il doit délivrer le solvant à un débit extrêmement constant et aussi reproductible que possible pour pouvoir convertir précisément les temps d’élution en volumes d’élution et utiliser avec précision la courbe d’étalonnage. Dans les conditions classiques, le débit est de l’ordre de 1 mL/min.
Injecteur
Le système de pompage est suivi d’un injecteur pour permettre l’introduction des échantillons. Le système le plus utilisé est l’injecteur à boucle qui permet d’injecter, sous haute pression, des quantités très reproductibles. Pour des colonnes classiques (30 cm de long et 7,2 mm de diamètre interne), le volume injecté ne doit pas dépasser 100 μL par colonne pour ne pas détruire les performances de la colonne (évaluée par son nombre de plateaux théoriques).
Colonne
Tous les composants de l’appareil de SEC sont reliés par des connexions à faible volume mort et des tubes capillaires (en acier inoxydable ou en plastique) pour minimiser l’élargissement des pics dû à l’appareillage.
L’efficacité des colonnes (évaluée en nombre de plateaux théoriques) peut être améliorée en augmentant la température, ce qui réduit aussi la pression dans la colonne.
Détecteurs
Les détecteurs utilisé en CES peuvent être classés en deux groupes :
- les détecteurs qui donnent des résultats proportionnels à la concentration du polymère dans la solution : réfractomètre différentiel, détecteurs par spectroscopie d'absorption et détecteurs de masse volumique ;
- les détecteur qui donnent des résultats proportionnels à la masse molaire moyenne en masse des polymères : viscosimètre, osmomètre continu, détecteurs à diffusion de la lumière[1].
Réfractomètre différentiel
Contrairement à la chromatographie liquide classique, le détecteur le plus communément utilisé est le réfractomètre différentiel qui produit un signal proportionnel à la masse et donc à la concentration du polymère. Le détecteur mesure une différence en indice de réfraction entre une cellule étalon qui contient de l'éluant pur et une cellule de détection où s'écoule l'échantillon élué. La cellule de détection a habituellement un volume de 10 μL pour minimiser le remélangeage et l’élargissement de bande.
Ce détecteur n’est pas particulièrement sensible, l'incrément d'indice de réfraction pouvant être très faible pour certains polymères, mais il a l’avantage d’être non spécifique et universel. Le réfractomètre est également extrêmement sensible aux variations de température et, comme une sensibilité de l’ordre de 10–7 unité d’indice de réfraction est nécessaire pour détecter les solutés à la sortie des colonnes, la température doit être stabilisée très soigneusement.
Spectroscopie d'absorption
Le spectromètre UV est parfois utilisé seul comme détecteur de concentration, mais il est plutôt couplé au réfractomètre comme détecteur secondaire de concentration pour l’analyse de composition, en particulier dans le cas des copolymères. Le spectromètre UV doit être réglé à une longueur d’onde particulière, à laquelle le soluté absorbe et le solvant est transparent. Le polymère doit posséder un bon coefficient d’absorption pour y être détecté. D’autres spectromètres, comme le spectromètre IR (absorbant dans l’infrarouge), sont aussi utilisés, par exemple pour l’analyse des polyoléfines. Les cellules de ces détecteurs ont habituellement un volume d’environ 10 μL
Viscosimètres
La détection viscométrique est particulièrement adaptée à l’utilisation de l’étalonnage universel car elle fournit une information de viscosité intrinsèque, nécessaire pour utiliser le paramètre • M. Au début, un viscosimètre Ubbelhode classique était simplement installé à la suite du réfractomètre. Ce montage n’est plus possible avec les colonnes modernes de SEC en raison de leur faible volume interne. En 1972, Oùano propose un autre type de viscosimètre fondé sur la mesure de la perte de charge dans un tube capillaire à l’aide d’un capteur de pression. Un viscosimètre, il a été amélioré depuis par l’utilisation d’un capteur de pression différentiel et d’un capillaire à faible volume mort.
À débit Q de solvant constant, la pression est directement proportionnelle à la viscosité. À viscosité constante, P est proportionnel au débit Q, ce qui donne un débitmètre extrêmement sensible. Par conséquent, le viscosimètre à simple capillaire, est très sensible aux fluctuations de débit et il est nécessaire d’utiliser un système de pompage renforcé avec un débit extrêmement stable débarrassé des à-coups de pression. Pour cette raison, ce viscosimètre peut être considéré comme un outil de diagnostic pour le système de pompage et même pour le reste de l’appareil, en plus de son utilisation plus fondamentale avec l’étalonnage universel. La détection viscosimétrique est aussi essentielle pour l’étude des ramifications longues dans les polymères. Ce sont des viscosimètres différentiels qui comportent plusieurs capillaires et plusieurs capteurs de pression et utilisent un volume de délai pour permettre une compensation de débit. Dans ce cas, le signal viscosimétrique différentiel est insensible aux variations de débit mais le signal réel doit être reconstruit à l’aide d’une mesure annexe de la ligne de base.
Détecteurs par diffusion de la lumière
C’est la technique LALLS (Low Angle Laser Light Scattering), qui consiste à mesurer la lumière diffusée à angle très faible (environ 5 degrés). Par conséquent, on peut admettre que le facteur de forme est très voisin de l’unité et que le signal est directement proportionnel à la masse moléculaire moyenne en poids. L’intérêt majeur de ce détecteur est qu’il ne nécessite pas d’étalonnage préalable du jeu de colonnes puisqu’il donne directement la masse moléculaire en fonction du volume d’élution. Lorsque le détecteur par diffusion de la lumière est utilisé en même temps qu’une courbe d’étalonnage en masse moléculaire, il peut donner des informations sur une élution anormale du polymère ou sur les ramifications longues. Récemment, une autre technologie a fait son apparition, le MALLS (Multi Angle Laser Light Scattering). Cet appareillage mesure simultanément la lumière diffusée suivant 16 angles différents (typiquement entre 20 ° et 150 °), et il est nécessaire d’extrapoler à angle nul, à l’aide d’un logiciel approprié, pour obtenir l’information de masse moléculaire. Comme l’appareil fonctionne à plusieurs angles, il donne, en plus, une mesure absolue du rayon de giration.
Osmomètre continu
Plus récemment, Yau a décrit un autre type de détecteur de masse, l’osmomètre continu. Ce détecteur est fondé sur la mesure de la perte de charge dans une petite colonne capillaire remplie de petites billes de gel gonflées par le solvant. La variation de pression osmotique provoque une variation du gonflement du gel quand le polymère est présent dans la colonne et, par conséquent, une variation de la perte de charge. Le principe de la mesure est le même que celui du viscosimètre différentiel de Yau. Ce détecteur est sensible à la masse moléculaire moyenne en nombre, puisqu’il s’agit d’osmométrie, et doit être très efficace dans le domaine des petites masses moléculaires. La relation entre la réponse du détecteur et la masse moléculaire absolue n’a pas encore été clairement établie et ce détecteur n’a pas encore connu de développement commercial.
Système d'acquisition
Le but principal d’une analyse SEC est de déterminer la courbe de distribution des masses moléculaires et d'évaluer les masses moléculaires moyennes. La procédure mathématique est relativement simple et peut être effectuée à partir de l’enregistrement graphique des chromatogrammes sur un enregistreur potentiométrique. Lorsque des calculs plus complexes sont nécessaires, principalement en SEC multidétection, on a besoin de méthodes de calculs informatiques plus compliquées et les méthodes simples ne sont plus applicables. Les détecteurs sont connectés à cet ordinateur par l’intermédiaire d’une interface qui assure la conversion analogique-numérique des signaux électriques. Les données SEC sont enregistrées sur une mémoire de stockage pour un traitement ultérieur à l’aide d’un logiciel SEC approprié.
Conditions expérimentales
Choix de la phase stationnaire et de l'éluant
Le choix du système phase stationnaire/éluant dépend principalement de la solubilité de l’échantillon à analyser. La phase mobile (l'éluant) doit être un bon solvant du polymère (au sens thermodynamique du terme) pour minimiser les mécanismes parasites de non exclusion, mais ce doit être aussi un bon agent gonflant du gel pour éviter un dégonflement du gel et un effondrement de la colonne. Pour les polymères organiques classiques, les gels de polystyrène sont parfaitement adaptés et le solvant le plus utilisé est le tétrahydrofuranne (THF). Il dissout de nombreux polymères et est parfaitement compatible avec les gels de polystyrène. Il peut néanmoins être remplacé par le toluène, le chloroforme ou le chlorure de méthylène. Malheureusement, quelques polymères industriels très importants ne sont pas solubles dans ces conditions et leur analyse doit être effectuée dans d’autres solvants, principalement à haute température. Les polyoléfines sont analysées dans l’o-dichlorobenzène ou le 1,2,4-trichlorobenzène à 135-145 oC, les polyamides dans le métacrésol ou dans l’alcool benzylique à 135 C°, les polyesters dans la N-méthylpyrrolidone et les polyuréthannes dans le diméthylformamide.
Les polymères hydrosolubles sont évidemment analysés en utilisant l’eau comme phase mobile. Quand le polymère est chargé électriquement(polyélectrolyte), des sels doivent être additionnés à la phase mobile pour en augmenter la force ionique et écranter les forces répulsives qui produisent l’effet polyélectrolyte bien connu. Des gels de silice poreuse dont la surface a été greffée chimiquement sont souvent utilisés mais il existe maintenant des gels polymères hydrophiles poreux très efficaces.
Choix de la température
La température est un paramètre important en SEC car elle affecte le mécanisme de séparation et la résolution de pics voisins. Idéalement, il est recommandé d'utiliser un four pour maintenir la température d'injection constante et ce, afin d'injecter dans des conditions stables et reproductibles. Il peut être nécessaire d’opérer à température élevée pour abaisser la viscosité de l'éluant qui pourrait être destructive pour l'échantillon de polymères et pour faciliter l’équilibre d’exclusion stérique.
De plus, certains polymères comme les polyoléfines ne possèdent pas de solvants propres à température ambiante et nécessitent d’être analysés à haute température à l’aide d’appareillages dédiés.
Choix du volume d'injection
Le choix du volume d'injection est un compromis entre la qualité de la détection et l'élargissement de bande. En effet, le réfractomètre différentiel étant un détecteur en masse, il peut être utile d'augmenter le volume d'injection pour ainsi augmenter la concentration. Une trop forte concentration de l'échantillon entraîne toutefois un élargissement de bande, qui se caractérise par une anomalie de séparation des faibles masses molaires (souvent visibles par un épaulement sur les chromatogrammes) et une diminution du volume d'élution.
Le domaine de températures possibles est aussi imposé par le solvant utilisé.
Choix du débit
Le choix du débit est également important dans la mesure où un débit trop faible crée un élargissement de bande et un débit trop fort peut favoriser des clivages dans les macromolécules, particulièrement dans le cas des polymères hyperbranchés. La valeur classique de débit est de 1 mL/min.
Analyse des polymères
Le premier niveau d’interprétation des données SEC est l’analyse de l’enregistrement des chromatogrammes sur un enregistreur potentiométrique. De nombreuses informations peuvent être obtenues en regardant juste les pics de polymère qui sont l’image de la distribution des volumes hydrodynamiques. Pour des opérations de contrôle, les chromatogrammes peuvent être considérés comme des empreintes digitales du polymère qui sont rapides à superposer pour effectuer une comparaison. La forme de la distribution (symétrique, non symétrique, monomodale, bimodale,…) caractérise la fonction de distribution du polymère en première approximation et la largeur de pics donne une idée de la polydispersité du polymère. Le volume d’élution au sommet du pic peut être mesuré facilement et donner une évaluation de la masse moléculaire moyenne par comparaison avec une courbe d’étalonnage, préalablement établie avec des standards de distribution étroite. Quand on a besoin d’informations quantitatives, il est nécessaire de mettre en œuvre un deuxième niveau d’analyse des données en utilisant des méthodes d’intégration de pic.
La SEC peut également être utilisée pour les composés de faible masse moléculaire (< 1 000 Dalton), des phases stationnaires avec des tailles de pore très petites étant disponibles. Puisque ces gels sont moins réticulés que les gels normaux, ils sont plus sensibles aux problèmes de gonflement, ce qui introduit des difficultés quand il s’agit de changer de solvant. Dans ce cas, les volumes d’élution sont moins sous le contrôle du mécanisme d’exclusion stérique et la relation entre le volume hydrodynamique et la masse moléculaire est moins bien définie et dépend de la structure moléculaire. En plus, à cause du comportement particulier du gel gonflé, le mécanisme de partage liquide/liquide entre la phase mobile et la phase stationnaire gonflée peut se produire et perturber les volumes d’élution. Par conséquent, la détermination des masses moléculaires dans ce domaine doit être conduite avec beaucoup de précautions, même si les conditions expérimentales semblent bien contrôlées. Cependant, des séparations qualitatives dans le domaine des faibles masses moléculaires peuvent être particulièrement utiles, par exemple pour la séparation des additifs dans les matières plastiques. Dans certains cas, cette méthode peut être utilisée d’une manière quantitative car ces types de pic sont très bien séparés du pic de polymère. Quand le jeu de colonnes est efficace exclusivement dans le domaine des faibles masses moléculaires, le but principal de l’expérience de SEC est d’énumérer le nombre de composants du mélange et d’effectuer une analyse quantitative d’au moins l’un d’entre eux. Ce type d’expérience est plus proche de la chromatographie liquide traditionnelle que de la SEC, sauf qu’ici tous les composés sont élués entre deux limites bien déterminées (V0 et V0 + Vp). Cette propriété est utilisée pour obtenir rapidement, par SEC, une estimation de la composition d’un mélange complexe, sans aucune optimisation de la phase mobile. Lorsque l’on a besoin d’une résolution très élevée pour la séparation de deux molécules très semblables, par exemple des isomères on peut augmenter énormément la résolution du système chromatographique en utilisant la technique du « recyclage » qui consiste à réintroduire automatiquement les éluats dans la pompe et la colonne à l’aide d’une vanne située à la sortie du détecteur. Cette méthode revient à accroître arbitrairement le nombre de colonnes et permet d’atteindre des efficacités extrêmement élevées.
SEC préparative
La SEC étant une technique non destructive, on peut l'utiliser comme méthode préparative pour récolter les éluats à la sortie du détecteur. Il suffit alors d’évaporer le solvant ou de précipiter le polymère pour récupérer les fractions collectées. Cependant, la quantité d’échantillon est extrêmement faible en SEC analytique et, pour purifier une quantité appréciable de produit, on doit utiliser un appareillage spécial muni d’une colonne de large diamètre. Un objectif de la SEC préparative est de purifier le polymère par la technique dite heart-cut qui consiste à collecter seulement le centre de la distribution et de rejeter les pieds qui contiennent les composants de plus faibles et de plus fortes masses moléculaires. Cette technique produit un matériau mieux défini et avec une plus faible polydispersité. Le fractionnement peut également être effectué de manière plus serrée pour préparer des composés bien définis en coupant la distribution en plusieurs fractions étroites. La purification des petites molécules, utilisant des gels de faibles porosités, peut également se faire en augmentant la quantité injectée pour obtenir des quantités appréciables de produit purifié, mais, industriellement, ce type de purification est plutôt réservé aux substances biologiques en raison de son coût très élevé[2].
Références
- Trathnigg, B. Determination of MWD and Chemical Composition of Polymers by Chromatographic Techniques. Prog. Polym. Sci. 1995, 20, 615-650. DOI 10.1016/0079-6700(95)00005-Z
- James Lesec, Chromatographie par perméation de gel - Chromatographie d’exclusion stérique, PE 1 465, Techniques de l’Ingénieur, traité Analyse et Caractérisation