Basic Local Alignment Search Tool
BLAST (acronyme de basic local alignment search tool) est une méthode de recherche heuristique utilisée en bioinformatique. Il permet de trouver les régions similaires entre deux ou plusieurs séquences de nucléotides ou d'acides aminés, et de réaliser un alignement de ces régions homologues.

| Développé par | Altschul S.F., Gish W., Miller E.W., Lipman D.J., NCBI |
|---|---|
| Dernière version | 2.9.0+ () |
| Écrit en | C++ et C |
| Système d'exploitation | Type Unix, Linux, macOS et Microsoft Windows |
| Environnement | Multiplate-forme |
| Formats lus | XML BLAST Output (d) |
| Formats écrits | XML BLAST Output (d) |
| Type | Outil bioinformatique |
| Licence | Domaine public |
| Site web | blast.ncbi.nlm.nih.gov |
Étant donné une séquence introduite par l'utilisateur, BLAST permet de retrouver rapidement dans des bases de données, les séquences répertoriées ayant des zones de similitude avec la séquence d'entrée[1]. Cette méthode est utilisée pour trouver des relations fonctionnelles ou évolutives entre les séquences et peut aider à identifier les membres d'une même famille de gènes.
Historique
Ce programme a été développé par Stephen Altschul, Warren Gish et David Lipman au National Center for Biotechnology Information (NCBI). La publication originale parue en octobre 1990, « Basic local alignment search tool »[2], a été citée plus de 90,000 fois[3], ce qui en fait l'une des plus citées dans le monde scientifique.
Principe
BLAST recherche dans une base de données de séquence des segments qui sont localement homologues à une séquence-test fournie par l'utilisateur (query sequence). BLAST utilise une matrice de similarité pour calculer des scores d'alignement. Il fournit un score pour chaque alignement trouvé et utilise ce score pour donner une évaluation statistique de la pertinence de cet alignement (probabilité qu'il soit dû au hasard).
Le principe de fonctionnement de BLAST, peut se décomposer en trois étapes[4] :
- la décomposition de la séquence-test en segments de longueur k (k-uplets) chevauchants et la recherche pour chacun d'eux de tous les k-uplets possibles ayant un score d'homologie supérieur à un seuil donné. BLAST constitue ainsi un dictionnaire de tous les k-uplets donnant une homologie locale minimum ;
- le balayage de la banque avec le dictionnaire ainsi constitué. Chaque fois que BLAST identifie une coïncidence dans la banque, il tente d'étendre l'homologie en amont et en aval du k-uplet initialement trouvé ;
- après extension de l'homologie, il évalue à partir du score obtenu la probabilité que celle-ci soit due au hasard (ou plus exactement, son espérance mathématique).
Création du dictionnaire de k-uplets
La séquence analysée est d'abord découpée en k-uplets chevauchants. Pour une séquence de protéine, on utilise typiquement des quadruplets d'acides aminés. Un segment de séquence comme FATCATY est par exemple découpé en :
FATC, ATCA, TCAT, CATY
Chacun de ces k-uplets est ensuite analysé pour identifier tous les k-uplets possibles qui donneraient un score d'alignement supérieur à une valeur seuil, fixée par l'utilisateur. Pour calculer ce score d'alignement, on utilise une matrice de similarité, M(a,b), souvent BLOSUM62 (avec a et b les deux acides aminés comparés). Par exemple, si on reprend le quadruplet FATC évoqué ci-dessus, le score qu'on obtient en l'alignant avec lui-même est de 24 si on utilise la matrice BLOSUM62 :
score(FATC,FATC) = M(F,F) + M(A,A) + M(T,T) + M(C,C) = 6 + 4 + 5 + 9 = 24
Quelques autres quadruplets donnent de bons scores d'alignement avec FATC, par exemple YATC, FASC ou FSTC, qui donnent des scores d'alignement supérieurs à 20 :
score(FATC,YATC) = 21 ; score(FATC,FASC) = 20 ; score(FATC,FSTC) = 21
En revanche, la grande majorité des autres quadruplets donne des scores d'alignements très mauvais, le plus souvent négatifs. Si on fixe un seuil élevé, comme 18 ou 20 dans notre exemple, on n'aura donc que quelques quadruplets possibles qui vont donner un score d'alignement supérieur à ce seuil.
BLAST va donc, pour chaque k-uplet de la séquence analysée, constituer un dictionnaire de tous les k-uplets possibles donnant un score supérieur au seuil. Le dictionnaire va également indiquer la position dans la séquence de référence où se trouve le k-uplet d'origine. Une fois constitué, ce dictionnaire contient la liste de tous les k-uplets possibles qui permettent d'obtenir un score initial d'alignement supérieur au seuil.
Recherche et extension de l'homologie
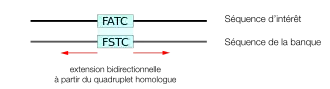
Une fois construit le dictionnaire de tous les k-uplets présentant un score seuil d'homologie avec la séquence de référence, BLAST va balayer la banque de séquences à analyser, en regardant successivement tous les k-uplets qui composent chaque séquence. À chaque fois, il vérifie si le k-uplet de la banque est présent dans le dictionnaire. S'il n'en fait pas partie, il passe au suivant. S'il est présent dans le dictionnaire, cela signifie qu'il existe un embryon de région homologue entre la séquence de référence et la séquence de la banque en cours d'analyse. Cette homologie présente un score minimum égal au seuil fixé dans la première étape.
BLAST va alors essayer de voir si cette région homologue s'étend au-delà du k-uplet de départ. Il va alors essayer d'étendre en amont et en aval la région pour voir si le score d'homologie augmente avec cette tentative d'extension. Si les deux séquences présentent effectivement une homologie locale autour du k-uplet de départ, l'extension va conduire à une augmentation effective du score, car de nouveaux acides aminés vont se trouver alignés. Si au contraire la tentative d'extension ne permet pas d'augmenter le score, parce que l'homologie ne continue pas, BLAST s'arrête. Si le score final après extension est supérieur à un seuil donné, l'alignement est conservé pour l'analyse finale.
Ce processus est effectué pour tous les k-uplets des séquences présentes dans la banque analysée, qui est balayée séquentiellement.
Analyse du score et évaluation de la pertinence
BLAST et ses versions dérivées (voir ci-dessous) est utilisé pour rechercher des séquences homologues à une séquence d'intérêt, introduite par l'utilisateur, à l'intérieur d'une banque de données de séquence. Ces banques de données, comme GenBank ou UniProt, sont très grandes, contenant typiquement plusieurs centaines de millions de séquences. La recherche exhaustive avec BLAST retourne en général plusieurs dizaines d'alignements avec la séquence d'intérêt. La question qui est alors posée est celle de la pertinence biologique de ces alignements : l'alignement est il simplement le résultat du hasard, parce qu'on a analysé un très grand nombre de séquences, ou bien est-il le reflet d'une véritable conservation biologique ?
Pour cela, au cours de la recherche, BLAST effectue une analyse de la distribution des scores d'alignement entre la séquence d'intérêt et la banque. Il ajuste cette distribution à une fonction de densité théorique, ce qui lui permet de calculer la probabilité et l'espérance mathématique de trouver un alignement donnant un score donné dans la banque, uniquement du fait du hasard. Les paramètres de cette fonction de densité varient en fonction des compositions en nucléotides ou acides aminés de la séquence et de la banque analysée.
Typiquement, BLAST va indiquer, pour chaque alignement, la valeur de cette espérance appelée E-value. Pour des alignements biologiquement pertinents, la E-value prend des valeurs infinitésimales (de 10−10 à 10−200), ce qui signifie qu'il est hautement improbable que le score d'alignement obtenu soit le fait du hasard.

Variations
Des données utilisées
Le terme blast peut être modifié en fonction de la nature de la séquence d'entrée, et de la base de données utilisée :
- blastn, de nucléotides, séquence nucléotidique contre une base de données de séquences nucléotidiques ;
- blastp, de protéines, séquence de protéine contre une base de données de séquences de protéines ;
- blastx, séquence nucléotidique traduite en séquence de protéine contre une base de données de séquences de protéines ;
- tblastn, séquence de protéine contre une base de données de séquences nucléotidiques traduites en séquences de protéines ;
- tblastx, séquence nucléotidique traduite en séquence de protéine contre une base de données de séquences nucléotidiques traduites en séquences de protéines.
De l'algorithme
Depuis sa création, différentes versions de l'algorithme ont été développées :
- BlastN, blast de séquences nucléotidiques, lent mais permet de retrouver des similarité localisées uniquement sur une partie des séquences ;
- BlastP, blast de séquences de protéines ;
- Megablast, rapide, permet de retrouver des séquences hautement similaires ;
- PSI-Blast (position-specific iterated BLAST), Blast relancé plusieurs fois par itération. À chaque itération une séquence consensus est déterminée à partir des résultats, et utilisée comme séquence source pour l'itération suivante ;
- PHI-BLAST (pattern hit initiated Blast), programme utilisant comme source une séquence protéique et un motif, celui-ci étant utilisé comme point de départ des recherches de similarité avec les séquences présentes dans les bases de données.
Notes et références
- Greg Gibson, Spencer V. Muse, Lionel Domenjoud, Raymond Cunin (trad. Lionel Domenjoud), Précis de génomique, Bruxelles/Paris, De Boeck Université, , 347 p. (ISBN 2-8041-4334-1), « 2 »
- (en) SF Altschul, W Gish, W Miller, EW Myers et DJ Lipman, « Basic local alignment search tool », Journal of molecular biology, vol. 215, no 3, , p. 403-10 (PMID 2231712, résumé)
- 20 000 fois
- Frédéric Dardel et François Képès, Bioinformatique. Génomique et post-génomique, Palaiseau, Éditions de l’École Polytechnique, , 246 p. (ISBN 2-7302-0927-1, présentation en ligne)
Voir aussi
Articles connexes
Liens externes
- (en) Site officiel
- (en) « mpiBLAST Demo »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) - mpiBLAST Parallel Version