RMN du proton
La RMN du proton est la spectroscopie RMN du proton (1H), un isotope de l'hydrogène. L'analyse d'un spectre RMN du proton peut se situer n'importe où entre très simple et très compliqué selon la structure de la molécule et des informations que l'on cherche à obtenir.
| Proton | |
| 1H | |
| Général | |
|---|---|
| Nombre de neutrons | 0 |
| Nombre de protons | 1 |
| Spin à l'état fondamental |
1/2 |
| Abondance naturelle [%] |
|
| Référence(s) | tétraméthylsilane (TMS) interne |
Méthodologie
L'analyse d'un spectre RMN (Résonance magnétique nucléaire) du proton fait appel à au moins trois opérations :
- L'attribution des résonances en fonction des déplacements chimiques (détermination des groupes fonctionnels)
- L'intégration des différentes résonances ou groupes de résonances (détermination de la proportion de chaque groupe)
- La détermination de la multiplicité du signal et éventuellement des constantes de couplages (détermination du nombre de voisins de chaque noyau / détermination de la structure)
Si cela n'est pas suffisant, il faut faire appel à la RMN de corrélation pour étudier les interactions entre les différents spins.
Un cas simple
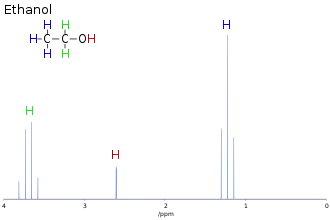
La détermination du déplacement chimique permet de déduire une grande partie des informations structurales sur la molécule étudiée. Par exemple, le spectre RMN de l'éthanol (CH3CH2OH), montre trois groupes de signaux spécifiques à trois déplacements chimiques différents (3,8 ; 2,6 et 1,3 ppm). La comparaison de ces déplacements avec des tables de déplacements chimiques[1] permet d'attribuer les trois signaux aux groupes CH2, OH et CH3, respectivement.
L'analyse de l'aire des différents groupes permet de confirmer cette attribution, puisqu'ils sont approximativement dans le rapport 2:1:3. Les logiciels d'analyse moderne permettent l'intégration des différentes résonances, mais ce paramètre doit être utilisé avec précaution, car de nombreux paramètres peuvent influencer l'intensité des résonances surtout dans les expériences complexes faisant intervenir plusieurs impulsions.
Une des informations les plus utiles en spectroscopie RMN pour la détermination de la structure de molécules organiques vient du couplage scalaire (ou couplage J, un cas particulier du couplage dipolaire) entre noyaux de spin non nul. Ce couplage résulte de l'interaction de différents états de spin à travers les liaisons chimiques d'une molécule. Il s'accompagne de l'éclatement des signaux de RMN en multiplets dépendant du nombre de voisins (de spin non nul). Ce couplage fournit un aperçu détaillé de la connectivité des atomes dans une molécule.
L'analyse des multiplets peut être parfois simple, notamment si les couplages sont du premier ordre):
- Si un noyau est couplé à noyaux de spin 1/2, sa résonance se divise en un multiplet composé de avec des ratios d'intensité qui peuvent être déterminé assez souvent suivant le triangle de Pascal :
- S’il y a d'autres couplages avec des noyaux non équivalents aux premiers, cela amènera d'autres séparations. Si le couplage a lieu entre des noyaux qui sont chimiquement équivalents (c'est-à-dire qui ont le même déplacement chimique), cela n'a pas d'effet sur les spectres RMN.


Par exemple, dans le spectre de protons de l'éthanol, le groupe CH3 est divisé en un triplet avec un rapport d'intensité de 1:2:1 par les deux voisins protons CH2. De même, le CH2 est divisé en un quartet avec un ratio d'intensité de 1:3:3:1 par les trois voisins protons CH3. En principe, les deux protons CH2 devraient être également divisés à nouveau en un doublet pour former un doublet de quartets à cause du couplage avec le proton hydroxyle, mais l'échange intermoléculaire du proton hydroxyle acide entraîne l'annulation de ce couplage.
La description ci-dessus suppose que la constante de couplage est faible en comparaison avec la différence de fréquences de déplacement chimique entre les spins non équivalents. Sinon, les intensités des multiplets sont modifiées et donc moins faciles à analyser (surtout si plus de deux spins sont impliqués).
{kind=link}