Maladie d'Ollier
La maladie d'Ollier ou enchondromatose est une maladie constitutionnelle de l'os[1] non héréditaire et sporadique, dans laquelle se développe des tumeurs cartilagineuses bénignes, les enchondromes. Ces derniers peuvent être de nombre, de taille, et de localisations différents expliquant l'extrême variabilité clinique entre les porteurs de cette maladie. Les principaux troubles observables sont une asymétrie et un raccourcissement des membres touchés, avec des déformations. Les symptômes apparaissent généralement pendant la petite enfance, lors de la croissance. Les atteintes sont généralement prédominantes d'un côté.
| Spécialité | Génétique médicale |
|---|
| Prévalence | 1/100 000 |
|---|
| CIM-10 | Q78.4 |
|---|---|
| CIM-9 | 756.4 |
| OMIM | 166000 |
| DiseasesDB | 9212 |
| MedlinePlus | https://medlineplus.gov/genetics/condition/ollier-disease/ |
| MeSH | D004687 |
![]() Mise en garde médicale
Mise en garde médicale
De nombreux patients atteints par la maladie d'Ollier sont susceptibles de développer des tumeurs malignes, en particulier des sarcomes osseux de type chondrosarcome, mais également des tumeurs dans d'autres tissus[2].
Un lien a été démontré entre la maladie d'Ollier et des mutations sur les gênes IDH1, IDH2[3] et PTH1R[4].
Actuellement, il n'existe aucun traitement de fond pour la maladie d'Ollier, mais les complications telles que les fractures, différences de longueur, ou tumeurs malignes agressives peuvent être traitées par chirurgie et/ou thérapie adjuvante.
L'association de la maladie d'Ollier à des hémangiomes prend le nom de syndrome de Maffucci[5].
Historique et épidémiologie
Connue depuis longtemps, ses caractéristiques ont été définies en 1899[6] par le chirurgien Louis Léopold Ollier qui lui donna son nom.
Sa prévalence est estimée à 1 cas sur 100 000 individus.
Cette maladie fait partie des maladies rares (117 personnes recensées en France en 2017 par l'Association Ollier-Maffucci Europe).
Synonymes : dyschondroplasie, hémichondrodystrophie d'Ollier, hémichondrodysplasie, chondromatose enchondrale multiple, syndrome d'Ollier-Mollin[7].
Diagnostic
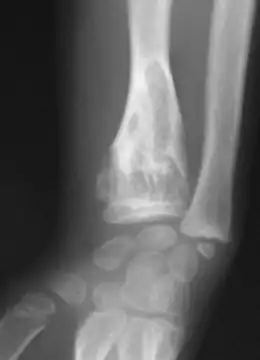
Les premières manifestations de la maladie apparaissent en général durant la petite enfance du fait d'un développement perturbé de l'os par la croissance d'enchondromes multiples entraînant déformations, déviations axiales et différences de longueur entre les membres. Les tuméfactions osseuses se présentent radiologiquement comme des lacunes claires arrondies, finissant par se calcifier[7]. La pose du diagnostic se fait par un examen radiologique.
Complications associées
La principale complication de la maladie d'Ollier est la transformation sarcomateuse d'un chondrome en chondrosarcome. Cette transformation est estimée, selon les études, en général entre 25 et 30% mais pouvant aller jusqu'à 50%[8]. Bien que le chondrosarcome soit la forme la plus courante de tumeur osseuse maligne secondaire dans les cas de maladies d'Ollier, d'autres formes comme le chordome et l'ostéosarcome peuvent se produire.
La maladie d'Ollier comporte également un risque élevé de favoriser le développement de tumeurs malignes agressives dans d'autres tissus[2] tel que le cerveau[9], les ovaires[10], ou le foie[11]. Il a été également rapporté des cas de maladie d'Ollier associés à des leucémies[12] ou myélodysplasies[13]. Une majorité de ces tumeurs comportent des mutations sur les gênes IDH1 et IDH2, pouvant expliquer le lien avec la maladie.
Non traitées, ces transformations malignes peuvent entraîner des conséquences fatales.
Traitement et suivi
Il n'existe pas de traitement de fond, la prise en charge est uniquement symptomatique. Selon le degré d'atteinte, les traitements seront essentiellement correctifs, tels que des allongement de membres (Illizarov/Fixateur externe), couplés à une prise en charge de la douleur. Les atteintes des chondromes évoluant avec la croissance, les effets de la maladie se stabilisent à la fin de la puberté, mais un suivi adulte régulier reste indispensable pour dépister précocement toutes formes de complications associées[14].
A l'âge adulte, le suivi clinique et radiologique sont les principaux outils pour dépister la survenue de transformation sarcomateuse des chondromes.
S'agissant du risque sur les autres tissus, il est recommandé d'effectuer un bilan complet (IRM cérébral[15], échographie hépatique, Imagerie gastro-intestinale) régulièrement[16].
Causes
Pendant de nombreuses années, la plupart des recherches n'ont pu identifier la cause de la maladie[17] - [18].
Les études récentes ont démontré que la plupart des cas (81%) de maladie d'Ollier sont causés par des mutations de l'isocitrate déshydrogénase 1 et 2 (IDH1 et IDH2)[3]. IDH1 et IDH2 sont normalement des enzymes qui catalysent la conversion de l'isocitrate en α-cétoglutarate. Les formes mutées d'IDH1 et d'IDH2 entraînent par la suite une conversion de l'α-cétoglutarate en 2-hydroxyglurate. La surproduction de 2-hydroxyglurate a pour effet d'inhiber la différenciation ostéogénique, tout en stimulant la différenciation chondrogénique, expliquant le développement des enchondromes[19] - [20].
Environ 8 à 10% des cas de maladie d'Ollier sont liés à une sur PTH1R[4].
Vu la distribution des atteintes sur un individu, il est hautement probable que la maladie résulte de mutations somatiques post-zygotique résultant un trouble génétique mosaïque[21].
Transmission
La maladie d'Ollier n'est pas transmissible et n'est pas une maladie héréditaire.
Les cas sont sporadiques et confirment la non-transmission mendélienne de cette maladie.
Références
- « Dictionnaire médical de l'Académie de Médecine », sur dictionnaire.academie-medecine.fr (consulté le )
- H. S. Schwartz, N. B. Zimmerman, M. A. Simon et R. R. Wroble, « The malignant potential of enchondromatosis », The Journal of Bone and Joint Surgery. American Volume, vol. 69, no 2, , p. 269–274 (ISSN 0021-9355, PMID 3805090, lire en ligne, consulté le )
- M. Fernanda Amary, Stephen Damato, Dina Halai et Malihe Eskandarpour, « Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2 », Nature Genetics, vol. 43, no 12, , p. 1262–1265 (ISSN 1546-1718, PMID 22057236, DOI 10.1038/ng.994, lire en ligne, consulté le )
- Alain Couvineau, Vinciane Wouters, Guylène Bertrand et Christiane Rouyer, « PTHR1 mutations associated with Ollier disease result in receptor loss of function », Human Molecular Genetics, vol. 17, no 18, , p. 2766–2775 (ISSN 1460-2083, PMID 18559376, PMCID 2722890, DOI 10.1093/hmg/ddn176, lire en ligne, consulté le )
- Di un caso encondroma ed ed angioma multiplo Movem Med Chir Napoli, 1881, 25, 399-412
- « De la dyschondroplasie » Bull Soc Chir Lyon 1899;3:22-27
- A. Manuila, Dictionnaire français de Médecine et de Biologie, t. 1, Masson, , p. 851.
- Jad M. El Abiad, Sarah M. Robbins, Bernard Cohen et Adam S. Levin, « Natural history of Ollier disease and Maffucci syndrome: Patient survey and review of clinical literature », American Journal of Medical Genetics. Part A, vol. 182, no 5, , p. 1093–1103 (ISSN 1552-4833, PMID 32144835, PMCID 8164175, DOI 10.1002/ajmg.a.61530, lire en ligne, consulté le )
- Charlotte Bonnet, Laure Thomas, Dimitri Psimaras et Franck Bielle, « Characteristics of gliomas in patients with somatic IDH mosaicism », Acta Neuropathologica Communications, vol. 4, , p. 31 (ISSN 2051-5960, PMID 27036230, PMCID 4818526, DOI 10.1186/s40478-016-0302-y, lire en ligne, consulté le )
- Pavlína Plevová et Hedvika Geržová, « Genetic Causes of Rare Pediatric Ovarian Tumors », Klinicka Onkologie: Casopis Ceske a Slovenske Onkologicke Spolecnosti, vol. 32, no Supplementum2, , p. 79–91 (ISSN 1802-5307, PMID 31409083, DOI 10.14735/amko2019S79, lire en ligne, consulté le )
- Rajalakshmi Govalan, Maha Guindi et Ju Dong Yang, « Liver Mass in a Young Male With Ollier Disease », Gastroenterology, vol. 161, no 5, , e4–e5 (ISSN 1528-0012, PMID 33812890, DOI 10.1053/j.gastro.2021.03.057, lire en ligne, consulté le )
- Matthew S. White, Paul L. Martin et Thomas W. McLean, « Acute myelogenous leukemia associated with Ollier disease », Pediatric Blood & Cancer, vol. 50, no 3, , p. 645–646 (ISSN 1545-5017, PMID 16991136, DOI 10.1002/pbc.21050, lire en ligne, consulté le )
- Mahwish Faizan, Sadia Anwar, null Nabeela et Agha Shabbir Ali, « Ollier's Disease with Myelodysplastic Syndrome », Journal of the College of Physicians and Surgeons--Pakistan: JCPSP, vol. 25, no 10, , p. 774–775 (ISSN 1681-7168, PMID 26454395, DOI 10.2015/JCPSP.774775, lire en ligne, consulté le )
- Avinash Kumar, Vijay Kumar Jain, Minakshi Bharadwaj et Rajendra Kumar Arya, « Ollier Disease: Pathogenesis, Diagnosis, and Management », Orthopedics, vol. 38, no 6, , e497–506 (ISSN 1938-2367, PMID 26091223, DOI 10.3928/01477447-20150603-58, lire en ligne, consulté le )
- Emmanuel Mandonnet, Philippe Anract, Elisabeth Martin et Thomas Roujeau, « Brain and skull base MRI findings in patients with Ollier-Maffucci disease: A series of 12 patient-cases », Clinical Neurology and Neurosurgery, vol. 160, , p. 147–151 (ISSN 1872-6968, PMID 28750360, DOI 10.1016/j.clineuro.2017.07.011, lire en ligne, consulté le )
- Jad M. El Abiad, Sarah M. Robbins, Bernard Cohen et Adam S. Levin, « Natural history of Ollier disease and Maffucci syndrome: Patient survey and review of clinical literature », American Journal of Medical Genetics. Part A, vol. 182, no 5, , p. 1093–1103 (ISSN 1552-4833, PMID 32144835, PMCID 8164175, DOI 10.1002/ajmg.a.61530, lire en ligne, consulté le )
- Twinkal C Pansuriya, Herman M Kroon et Judith VMG Bovée, « Enchondromatosis: insights on the different subtypes », International Journal of Clinical and Experimental Pathology, vol. 3, no 6, , p. 557–569 (ISSN 1936-2625, PMID 20661403, PMCID 2907117, lire en ligne, consulté le )
- (en) Saravanaraja Muthusamy, Sheila A. Conway et H. Thomas Temple, « Five Polyostotic Conditions That General Orthopedic Surgeons Should Recognize (or Should Not Miss) », Orthopedic Clinics of North America, vol. 45, no 3, , p. 417–429 (ISSN 0030-5898, DOI 10.1016/j.ocl.2014.04.004, lire en ligne, consulté le )
- Johnny Suijker, Hans J. Baelde, Helene Roelofs et Anne-Marie Cleton-Jansen, « The oncometabolite D-2-hydroxyglutarate induced by mutant IDH1 or -2 blocks osteoblast differentiation in vitro and in vivo », Oncotarget, vol. 6, no 17, , p. 14832–14842 (ISSN 1949-2553, PMID 26046462, PMCID 4558118, DOI 10.18632/oncotarget.4024, lire en ligne, consulté le )
- Yonghui Jin, Hassan Elalaf, Makoto Watanabe et Sakura Tamaki, « Mutant IDH1 Dysregulates the Differentiation of Mesenchymal Stem Cells in Association with Gene-Specific Histone Modifications to Cartilage- and Bone-Related Genes », PloS One, vol. 10, no 7, , e0131998 (ISSN 1932-6203, PMID 26161668, PMCID 4498635, DOI 10.1371/journal.pone.0131998, lire en ligne, consulté le )
- Char Loo Tan, Balamurugan Vellayappan, Bingcheng Wu et Tseng Tsai Yeo, « Molecular profiling of different glioma specimens from an Ollier disease patient suggests a multifocal disease process in the setting of IDH mosaicism », Brain Tumor Pathology, vol. 35, no 4, , p. 202–208 (ISSN 1861-387X, PMID 30159860, DOI 10.1007/s10014-018-0327-y, lire en ligne, consulté le )
Liens externes
- Association Ollier Maffucci Europe
- « Ollier disease », Orphanet Journal of Rare Diseases, vol. 1, no 37, (DOI 10.1186/1750-1172-1-37, lire en ligne)