Hélice-coude-hélice
Un domaine hélice-coude-hélice (HTH, ou Helix-Turn-Helix en anglais) est un élément de structure secondaire des protéines et un motif structurel important des protéines de liaison à l'ADN. Distinct du domaine hélice-boucle-hélice avec lequel il ne doit pas être confondu, il est formé de deux hélices α reliées par un court segment fléchi et est présent dans de nombreuses protéines intervenant dans la régulation de l'expression génétique[1]. Son identification résulte de l'observation de similitudes entre des gènes codant les protéines Cro, CAP (en) et répresseur λ de régulation de la transcription chez le phage λ et Escherichia coli, qui se trouvent posséder en commun une séquence de 20 à 25 résidus d'acides aminés facilitant la reconnaissance de l'ADN[2] - [3] - [4] - [5].
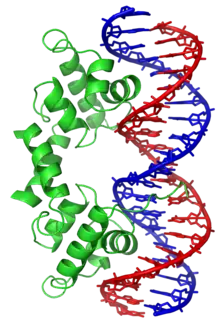
Structure et typologies
La structure hélice-coude-hélice est un motif de liaison à l'ADN. Les deux hélices α assurent la reconnaissance et la liaison à l'ADN, l'une à l'extrémité N-terminale et l'autre à l'extrémité C-terminale. Dans la plupart des cas, comme pour le répresseur Cro, c'est la seconde hélice α (côté C-terminal) qui intervient le plus dans la reconnaissance de l'ADN, de sorte qu'on l'appelle hélice de reconnaissance. Elle se lie au grand sillon de l'ADN à travers une série de liaisons hydrogène et d'interaction de Van der Waals avec les bases exposées à ce sillon. L'autre hélice α, côté N-terminal, stabilise l'interaction entre la protéine et l'ADN, mais ne joue pas de rôle particulier dans la reconnaissance de cet ADN[2]. Ces deux hélices partagent toujours la même orientation relative[6].
Il existe plusieurs types de motifs hélice-coude-hélice en fonction de leur structure et de l'arrangement tridimensionnel des hélices[6] - [7] - [8]. Certains sont décrits ci-dessous :
- À deux hélices — C'est le plus simple de ces motifs. Un homéodomaine enroulé comprenant deux hélices α et le coude a été identifié comme domaine protéique à repliement indépendant ultrarapide[9].
- À trois hélices — On en trouve un exemple dans l'activateur de transcription MYB (en)[10].
- À quatre hélices et plus — Le motif HTH à quatre hélices possède une hélice C-terminale de plus que le motif à trois hélices. On le trouve dans le domaine HTH de liaison à l'ADN de type LuxR (en) des facteurs de transcription bactériens et dans le motif HTH des répresseurs TetR (en)[11]. Il existe également des motifs HTH possédant encore davantage d'hélices[12].
- HTH « ailé » — le motif HTH ailé (wHTH, pour winged helix-turn-helix) est formé d'un faisceau de trois hélices α et d'un feuillet β à trois ou quatre brins formant un aile. La géométrie de ces éléments varie selon les différents motifs wHTH. Celui de la famille des facteurs de transcription ETS (en), par exemple, est formé d'un motif HTH et d'un feuillet β à quatre brins formant un échafaudage dans l'ordre α1-β1-β2-α2-α3-β3-β4, la troisième hélice étant celle de reconnaissance[13] - [14].
- Autres motifs HTH modifiés — Le domaine de liaison à l'ADN du régulateur de résistance aux antibiotiques multiple MarR présente un motif HTH ailé possédant une hélice C-terminale supplémentaire[15].
Références
- (en) R. G. Brennan et B. W. Matthews, « The helix-turn-helix DNA binding motif », Journal of Biological Chemistry, vol. 264, no 4, , p. 1903-1906 (PMID 2644244, lire en ligne)
- (en) B. W. Matthews, D. H. Ohlendorf, W. F. Anderson et Y. Takeda, « Structure of the DNA-binding region of lac repressor inferred from its homology with cro repressor », Proceedings of the National Academy of Sciences of the United States of America, vol. 79, no 5, , p. 1428-1432 (PMID 6951187, PMCID 345986, DOI 10.1073/pnas.79.5.1428, lire en ligne)
- (en) W. F. Anderson, D. H. Ohlendorf, Y. Takeda et B. W. Matthews, « Structure of the cro repressor from bacteriophage λ and its interaction with DNA », Nature, vol. 290, no 5809, , p. 754-758 (PMID 6452580, DOI 10.1038/290754a0, lire en ligne)
- (en) David B. McKay et Thomas A. Steitz, « Structure of catabolite gene activator protein at 2.9 Å resolution suggests binding to left-handed B-DNA », Nature, vol. 290, no 5809, , p. 744-749 (PMID 6261152, DOI 10.1038/290744a0, lire en ligne)
- (en) Carl O. Pabo et Mitchell Lewis, « The operator-binding domain of λ repressor: structure and DNA recognition », Nature, vol. 289, no 5873, , p. 443-447 (PMID 7088190, DOI 10.1038/298443a0, lire en ligne)
- (en) René Wintjens et Marianne Rooman, « Structural classification of HTH DNA-binding domains and protein-DNA interaction modes », Journal of Molecular Biology, vol. 262, no 2, , p. 294-313 (PMID 8831795, DOI 10.1006/jmbi.1996.0514, lire en ligne)
- (en) Masashi Suzuki et Steven E. Brenner, « Classification of multi-helical DNA-binding domains and application to predict the DBD structures of σ factor, LysR, OmpR/PhoB, CENP-B, Rap1, and XylS/Ada/AraC », FEBS Letters, vol. 372, nos 2-3, , p. 215-212 (PMID 7556672, DOI 10.1016/0014-5793(95)00988-L, lire en ligne)
- (en) L. Aravind, Vivek Anantharaman, Santhanam Balaji, M. Mohan Babu et Lakshminarayan M. Iyer, « The many faces of the helix-turn-helix domain: Transcription regulation and beyond », FEMS Microbiology Reviews, vol. 29, no 2, , p. 231-262 (PMID 15808743, DOI 10.1016/j.fmrre.2004.12.008, lire en ligne)
- (en) Tomasz L. Religa, Christopher M. Johnson, Dung M. Vu, Scott H. Brewer, R. Brian Dyer et Alan R. Fersht, « The helix–turn–helix motif as an ultrafast independently folding domain: The pathway of folding of Engrailed homeodomain », Proceedings of the National Academy of Sciences of the United States of America, vol. 104, no 22, , p. 9272-9277 (PMID 17517666, PMCID 1890484, DOI 10.1073/pnas.0703434104, lire en ligne)
- (en) K. Ogata, H. Hojo, S. Aimoto, T. Nakai, H. Nakamura, A. Sarai, S. Ishii et Y. Nishimura, « Solution structure of a DNA-binding unit of Myb: a helix-turn-helix-related motif with conserved tryptophans forming a hydrophobic core », Proceedings of the National Academy of Sciences of the United States of America, vol. 89, no 14, , p. 6428-6432 (PMID 1631139, PMCID 49514, DOI 10.1073/pnas.89.14.6428, lire en ligne)
- (en) W. Hinrichs, C. Kisker, M. Duvel, A. Muller, K. Tovar, W. Hillen et W. Saenger, « Structure of the Tet repressor-tetracycline complex and regulation of antibiotic resistance », Science, vol. 264, no 5157, , p. 418-420 (PMID 8153629, DOI 10.1126/science.8153629, lire en ligne)
- (en) Junji Iwahara et Robert T. Clubb, « Solution structure of the DNA binding domain from Dead ringer, a sequence‐specific AT‐rich interaction domain (ARID) », EMBO Press, vol. 18, no 21, , p. 6084-6094 (PMID 10545119, PMCID 1171673, DOI 10.1093/emboj/18.21.6084, lire en ligne)
- (en) L. W. Donaldson, J. M. Petersen, B. J. Graves et L. P. McIntosh, « Solution structure of the ETS domain from murine Ets-1: a winged helix-turn-helix DNA binding motif », The EMBO Journal, vol. 15, no 1, , p. 125-134 (PMID 8598195, PMCID 449924)
- (en) Andrew D. Sharrocks, A.Louise Brown, Yan Ling et Paula R. Yates, « The ETS-domain transcription factor family », The International Journal of Biochemistry & Cell Biology, vol. 29, no 12, , p. 1371-1387 (PMID 9570133, DOI 10.1016/S1357-2725(97)00086-1, lire en ligne)
- (en) Michael N. Alekshun, Stuart B. Levy, Tanya R. Mealy, Barbara A. Seaton et James F. Head, « The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 Å resolution », Nature Structural Biology, vol. 8, no 8, , p. 710-714 (PMID 11473263, DOI 10.1038/90429, lire en ligne)