Ataxie spinocérébelleuse
Les ataxies spinocérébelleuses constituent un groupe de maladies neurodégénératives très hétérogènes tant du point de vue clinique que génétique (caractère récessif ou caractère dominant, d’évolution lente et progressive) et dont l'issue est souvent fatale. Elles se caractérisent par un syndrome cérébelleux qui se traduit notamment par des troubles de la marche et de l'équilibre. Ces troubles sont la conséquence de la dégénérescence du cervelet et plus ou moins de ses afférences et efférences. Souvent, les individus n'ont pas conscience d'être porteurs du gène responsable, jusqu'à ce qu'ils aient des enfants qui commencent à montrer des signes cliniques de la maladie. De nombreuses formes d'ataxie spinocérébelleuse restent peu connues, et des études sont en cours afin de mieux les caractériser. Cette maladie est encore incurable (pas de traitement )
Causes
Les ataxies spinocérébelleuses surviennent à la suite d'une dégénérescence des cellules de la moelle épinière, mais aussi du cervelet. En effet, le cervelet assure le maintien de la posture, de l'équilibre, et coordonne les mouvements volontaires. Ainsi, lorsque le cervelet ne peut plus assurer ses fonctions, les symptômes propres aux ataxies apparaissent et évoluent de manière plus ou moins rapide.
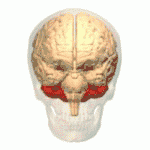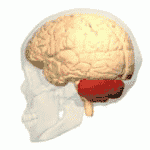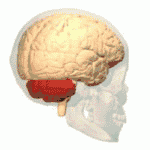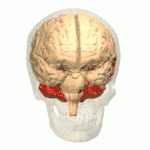
Au fil de l'évolution de la maladie, les muscles du sujet atteint répondent de moins en moins aux messages nerveux, ce qui donne lieu à des troubles moteurs comme l'hypotonie musculaire.
Du point de vue génétique, les causes diffèrent d'une ataxie spinocérébelleuse à l'autre.
Pour les ataxies spinocérébelleuses à transmission dominante, Margolis (2002) a proposé une classification des maladies en trois catégories : les maladies résultant de répétitions excessives de trinucléotides CAG ou de CAA codant la glutamine ; les maladies liées aux canaux ioniques du calcium ou du potassium ; et les maladies résultant de répétitions excessives de trinucléotides des régions non codantes de divers gènes[1].
Pour les ataxies spinocérébelleuses à transmission récessive, plusieurs classifications ont été proposées, mais aucune d'entre elles n'a fait l'objet d'un consensus. Cependant, plusieurs ataxies de ce type ont des points communs : un dysfonctionnement mitochondrial; un défaut de réparation des lésions de l'ADN; des troubles du métabolisme de l'ARN; un défaut d'assemblage des lipoprotéines; un dysfonctionnement des protéines chaperonnes, et des pathologies peroxysomales[2].
Quel que soit le mode de transmission de la maladie (récessif ou dominant), les altérations des gènes donnent lieu à des protéines défectueuses, qui elles-mêmes affecteront les cellules nerveuses (principalement celles du cervelet et de la moelle épinière).
Diagnostic
Le diagnostic des ataxies spinocérébelleuses se fait par plusieurs moyens. En plus de l'examen somatique, d'autres techniques seront utilisées afin de confirmer ou non la présence de la maladie. Parmi ces techniques, on trouvera l'IRM (notamment afin de vérifier l'aspect du cervelet), les tests biochimiques (sang et urine), et les tests génétiques (afin d'identifier un gène connu pour être responsable de la maladie). Les ataxies spinocérébelleuses étant très différentes les unes des autres sur le plan génétique, elles donnent ainsi lieu à des symptômes relativement divers.
Les ataxies spinocérébelleuses autosomiques dominantes (ACAD)
Le tableau ci-dessous répertorie les ACAD les plus fréquentes (SCA1, 2, 3, 6 et 7), avec leurs noms, l'âge auquel la maladie se manifeste, les symptômes et les gènes impliqués.
| Nom | Âge | Symptômes | Gène impliqué |
|---|---|---|---|
| SCA1 | Vers la trentaine (le plus souvent) | Manque de coordination des mains, troubles de l'équilibre lors de la marche, dysphagie, neuropathie, spasticité | ATXN1 |
| SCA2 | Vers la trentaine (le plus souvent) | Neuropathie, mouvements oculaires très lents, crampes musculaires, tremblements (parfois), dysphagie, troubles de l'élocution, spasticité, faiblesse générale et troubles de la mémoire | ATXN2 |
| SCA3 | Vers la trentaine (le plus souvent) | Diplopie, dysphagie, troubles de la marche, troubles de l'élocution, maladresse, ataxie progressive, hyperréflexie, nystagmus, paraplégie spastique | ATXN3 |
| SCA6 | Vers la quarantaine (le plus souvent) | Troubles de l'élocution, pertes d'équilibre, nystagmus, neuropathie, diminution du tonus musculaire | CACNA1A |
| SCA7 | L'âge de début varie (de l'enfance à la cinquantaine) : il est souvent plus précoce pour les générations suivantes. | Baisse progressive de l'acuité visuelle, mouvements oculaires lents, perte de contrôle moteur, troubles de l'élocution, dysphagie | ATXN7 |
Les ataxies spinocérébelleuses autosomiques récessives (ACAR)
Le tableau ci-dessous répertorie quelques ACAR, avec leurs noms, l'âge auquel la maladie se manifeste, les symptômes et les gènes impliqués. Les ACAR présentées ici peuvent être classées en trois groupes[3], à savoir : celles avec un syndrome cérébelleux pur (ARCA2 et MSS) ; celles avec un syndrome spino-cérébelleux, cordonal postérieur et une polyneuropathie sensitive (FRDA et SANDO) ; et celles avec un syndrome cérébelleux et une polyneuropathie sensitivo-motrice (AT et ARSACS).
| Nom | Âge | Symptômes | Gène impliqué |
|---|---|---|---|
| ARCA2 | Entre 1 et 11 ans | Hypotonie, chutes (inaugurales ou non), ataxie avec atrophie cérébelleuse, intolérance à l'exercice, réflexes ostéo-tendineux particulièrement vifs, pieds creux, épilepsie ou « stroke-like syndrome » | ADCK3 (CABC1) |
| MSS | Dès la naissance | Hypotonie, ataxie cérébelleuse, cataracte bilatérale, retard psychomoteur (modéré), retard de croissance staturo-pondéral, hypogonadisme, myopathie avec amyotrophie et retard moteur progressif | SIL1 |
| FRDA | Entre 2 et 60 ans (souvent à l'adolescence) | Ataxie cérébelleuse et proprioceptive, aréflexie ostéo-tendineuse, dysarthrie cérébelleuse, dysphagie, signe de Babinski bilatéral, déficit moteur, scoliose, cardiomyopathie hypertrophique, diabète, instabilité de la fixation oculaire et atrophie optique | FXN |
| SANDO | Entre 30 et 60 ans | Ophtalmoplégie, dysarthrie, dysphagie, équivalents migraineux, épilepsie, myopathie | POLG |
| AT | Avant l'âge de 5 ans | Infections récurrentes (surtout naso-sinusiennes et bronchopulmonaires), prédisposition accrue aux cancers, apraxie oculomotrice, ataxie cérébelleuse, mouvements choréo-dystoniques, télangiectasies | ATM |
| ARSACS | Avant l'âge de 12 ans | Paraparésie spastique, ataxie cérébelleuse, altération des capacités cognitives (parfois), atteinte oculaire et perte de la marche vers l'âge de 40 ans | SACS |
Épidémiologie
La fréquence des ataxies cérébelleuses autosomiques dominantes (ACAD) est d'environ 5 pour 100 000. Les ACAD touchent principalement les individus (hommes ou femmes) âgés de 25 à 50 ans[4]. La fréquence des ataxies cérébelleuses autosomiques récessives (ACAR), quant à elle, est d'environ 6 pour 100 000. Les ACAR se manifestent principalement avant l’âge de 20 ans (tant pour les hommes que pour les femmes), mais elles peuvent se manifester plus tardivement[5]. La plus fréquente des ACAR est l'ataxie de Friedreich.
Traitements
Il n'existe actuellement aucun traitement capable de guérir les ataxies spinocérébelleuses. Ainsi, les traitements actuels sont uniquement symptomatiques, comme la buspirone[6], qui aide à réduire les symptômes moteurs. La rééducation fonctionnelle, l'accompagnement médical (neurologue et médecin traitant), paramédical (Masseur-kinésithérapeute, ergothérapeute, orthophoniste...), psychologique et social sont donc essentiels pour aider le patient à vivre au mieux avec son handicap.
Culture populaire
Une adolescente japonaise de 15 ans, Aya Kitō, atteinte de cette maladie, laissa derrière elle un journal intime. Ce journal intime sortira au japon et se vendra à 18 millions d'exemplaires, à la suite de cela un film du nom de Ichi Rittoru no Namida sortira puis par la suite un drama (série japonaise) fut créé pour lui rendre hommage : One litre of tears. Depuis des personnes atteintes de la même maladie vont se recueillir sur sa tombe.
Articles connexes
Liens externes
Références
- Robert Lalonde et Catherine Strazielle, « La neurochimie du comportement dans les maladies neurodégénératives : hérédo-ataxies, Parkinson et Alzheimer », dans Thérèse Botez-Marquard et François Boller (dir.), Neuropsychologie clinique et neurologie du comportement, Troisième édition, Les Presses de l'Université de Montréal, , 849 p.
- Mathieu Anheim, « Les ataxies cérébelleuses autosomiques récessives », Revue neurologique, vol. 167, no 5, , p. 372–384
- Mathieu Anheim, « Les ataxies cérébelleuses autosomiques récessives », Neurologie.com, vol. 2, no 2, , p. 1–5
- Les ataxies cérébelleuses dominantes sur le site de l'Encyclopédie internationale multilingue de la réadaptation
- Aspects médicaux et scientifiques des syndromes cérébelleux, Association Connaître les Syndromes Cérébelleux, avril 2011
- Buspirone sur le site Medify.com