Maladie de Degos
La maladie de Degos (aussi appelée papulose atrophiante maligne) est une vasculopathie extrêmement rare qui affecte le revêtement intérieur des petites et moyennes veines, ce qui entraîne l'occlusion (blocage du vaisseau) et des infarctus du tissu.
| Spécialité | Cardiologie et dermatologie |
|---|
| CIM-10 | I77.89 |
|---|---|
| CIM-9 | 447.8 |
| OMIM | 602248 |
| DiseasesDB | 29425 |
| eMedicine | 1087180 |
| MeSH | D054853 |
![]() Mise en garde médicale
Mise en garde médicale
La maladie prend son nom de Robert Degos qui l’a reconnue comme une entité clinique en 1942, après qu'elle a été décrite, en premier, par l'anatomopathologiste autrichien Walter Köhlmeier en 1941[1].
Cause
Elle est inconnue. Il existe peut-être une participation génétique avec l'existence de formes familiales[2]. Il semble exister une dérégulation de la réponse à l'interféron-alpha[3].
Épidémiologie
Il y a moins de cinquante malades vivants, dans le monde à ce jour. Moins de 200 cas ont été rapportés dans la littérature médicale. Elle est plus fréquente chez l'adulte de type caucasien[4].
Description
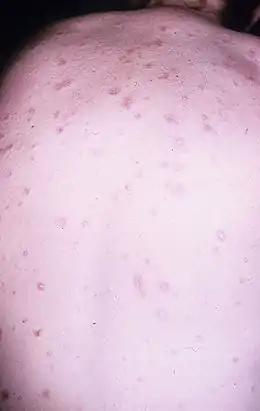
Les vaisseaux sanguins affectés sont ceux qui fournissent la peau, le tube digestif et le système nerveux. Cela conduit à une ischémie de l'intestin avec possible perforation[5], des lésions chroniques de la peau, lésions oculaires, accidents vasculaires cérébraux, lésions de la moelle épinière, mononévrite multiple (anciennement dénommée multinévrite), épilepsie, maux de tête ou troubles cognitifs. Des épanchements pleuraux ou péricardiques sont également signalés.
Sa similitude avec le lupus érythémateux disséminé[6] ou simplement cutanée[7] a été soulignée.
Évolution
Cette maladie peut-être fatale au bout de 2 ou 3 ans en moyenne, bien que dans certains cas, une forme bénigne puisse apparaitre (on parle alors d'acanthose de Degos) qui affecte uniquement la peau. Cette forme atteint plutôt les femmes, sans spécificité biologique[4].
Traitement
Les options de traitement sont limitées. Elles consistent principalement à des médicaments antiplaquettaires[8], anticoagulants ou des immunosuppresseurs[9]. Le traitement par antiagrégants plaquettaires permet parfois une rémission des signes cutanés, récidivant à leur arrêt[10].
Notes et références
- (en) « Article « Syndrome de Köhlmeier-Degos » », sur Who Named It?
- (en) Katz SK, Mudd LJ, Roenigk HH Jr, « Malignant atrophic papulosis (Degos' disease) involving three generations of a family » J Am Acad Dermatol. 1997;37:480-484.
- (en) Magro CM, Poe JC, Kim C et al. « Degos disease: a C5b-9/interferon-α-mediated endotheliopathy syndrome » Am J Clin Pathol. 2011;135:599-610.
- (en) Wilson J, Walling HW, Stone MS, « Benign cutaneous Degos disease in a 16-year-old girl » Pediatr Dermatol. 2007;24:18-24.
- (en) Amaravadi RR, Tran TM, Altman R, Scheirey CD, « Small bowel infarcts in Degos disease » Abdom Imaging 2008;33:196-199.
- (en) Ball E, Newburger A, Ackerman AB, « Degos' disease: a distinctive pattern of disease, chiefly of lupus erythematosus, and not a specific disease per se » Am J Dermatopathol. 2003;25:308-320.
- (en) Mutizwa MM, Tang MB, Ng SK, « Prominent Degos-like skin lesions in a patient with chronic cutaneous lupus erythematosus » Dermatol Online J. 2010;16:5-5.
- (en) Stahl D, Thomsen K, Hou-Jensen K, « Malignant atrophic papulosis: treatment with aspirin and dipyridamole » Arch Dermatol. 1978;114:1687-1689.
- (en) Gupta S, Dogra S, Saikia UN, Yadav S, Kanwar AJ, « Degos disease with dermatomyositis-like phenomenon: a diagnostic dilemma and a therapeutic challenge » J Cutan Med Surg. 2011;15:162-166.
- (en) Drucker CR, « Malignant atrophic papulosis: response to antiplatelet therapy » Dermatologica 1990;180:90-92.