Déficit en 3-hydroxyacyl-coenzyme A déshydrogénase des acides gras à chaîne longue
Le déficit en 3-hydroxyacyl-coenzyme A déshydrogénase des acides gras à chaîne longue, souvent abrégé en déficit en LCHAD, est l'une des maladies rares à transmission autosomique récessive lié à l'oxydation des acides gras[1] qui empêche le corps de convertir certaines graisses en énergie. Cela peut être dangereux, en particulier pendant les périodes de jeûne.
![]() Mise en garde médicale
Mise en garde médicale
Les symptômes
En général, les premiers signes et symptômes de ce trouble se produisent au cours de l'enfance et peuvent inclure des difficultés d'alimentation, un état léthargique, des hypoglycémie, une hypotonie, des problèmes de foie, et des anomalies de la rétine. Des douleurs musculaires, la dégradation du tissu musculaire, et des anomalies dans le système nerveux qui affectent les bras et les jambes (neuropathie périphérique) peuvent se produire plus tard dans l'enfance. Il existe aussi un risque de complications cardiaques sévères et des problèmes respiratoires, de coma et de mort subite. Les épisodes de déficit en LCHAD peuvent être déclenchés par des périodes de jeûne ou par des maladies telles que des infections virales.
La génétique
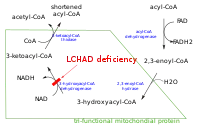
Des mutations dans le gène HADHA conduisent à une insuffisance des niveaux d'une enzyme appelée 3-hydroxyacyl-coenzyme A (CoA) déshydrogénase à longue chaîne, qui fait partie d'un ensemble complexe de protéines connues sous le nom de protéine trifonctionnelle mitochondriale. Les longues chaînes d'acides gras issues des aliments et la graisse du corps ne peuvent pas être métabolisées et traitées sans suffisamment de niveaux de cette enzyme. En conséquence, ces acides gras ne sont pas converties en énergie, ce qui peut conduire à des caractéristiques de ce trouble, comme la léthargie et l'hypoglycémie. Les acides gras à chaîne longue ou partiellement métabolisés en acides gras peuvent s'accumuler dans les tissus et causer des dommages au foie, au cœur, à la rétine et aux muscles, provoquant des complications plus graves.
Pronostic
En 2001, une étude de suivi sur 50 patients. De ces 38% sont morts dans l'enfance, tandis que le reste souffert de problèmes avec la morbidité[2].
Références
- (en) Genetics Home Reference, « LCHAD deficiency », sur Genetics Home Reference (consulté le )
- (en) Margarethe E. J. den Boer, Ronald J. A. Wanders, Andrew A. M. Morris et Lodewijk IJlst, « Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency: Clinical Presentation and Follow-Up of 50 Patients », Pediatrics, vol. 109, no 1, , p. 99–104 (ISSN 0031-4005, PMID 11773547, DOI 10.1542/peds.109.1.99, lire en ligne)