Cycloaddition 1,3-dipolaire
La cycloaddition 1,3-dipolaire, aussi connue sous le nom de cycloaddition de Huisgen ou réaction de Huisgen[1] - [2] est une réaction en chimie organique appartenant à la grande famille des cycloadditions péricycliques concertées. C'est une réaction entre un 1,3-dipôle et un dipolairophile, la plupart du temps un alcène substitué, pour former un cycle à cinq atomes. Rolf Huisgen fut le premier à effectuer des recherches sur cette réaction en variant les 1,3-dipôles et à considérer son importance dans la synthèse d'hétérocycles à cinq atomes.

Preuve du mécanisme
Firestone avait dans un premier temps tablé sur l'existence d'un intermédiaire diradical, mais ses travaux furent contredits par ceux de Huisgen, qui prévoyait un processus concerté, bien que cette hypothèse fut longtemps sujette à caution. C'est néanmoins aujourd'hui l'hypothèse de Huisgen qui est communément acceptée[3], soutenue en cela par plusieurs preuves d'un mécanisme concerté :
- accroitre le groupe carbonyle du 1,3-dipole décroit sa réactivité par stabilisation de son état fondamental ;
- les effets du solvant sur la réaction sont mineurs, or si la réaction se produisait via un intermédiaire chargé, on devrait avoir une augmentation de la vitesse de réaction avec la polarité du solvant ;
- la réaction se fait par cis-addition car deux liaisons sont formées simultanément, il n'y a donc pas d'intermédiaire qui pourrait effectuer une rotation et brouiller la stéréochimie du produit.
De nombreux autres calculs et expérimentations ont également été menés pour vérifier la nature concertée du mécanisme.
Réactivité
Un processus concerté tel qu'une cycloaddition 1,3 nécessite un état de transition hautement ordonné, mais seulement des besoins enthalpiques modérés. En utilisant des réactifs en compétition, des taux relatifs d'additions pour différentes cycloadditions ont été trouvés, ce qui permet de définir plusieurs facteurs de réactivité :
- la conjugaison, en particulier avec des groupes aromatiques, accroit la vitesse de réaction par stabilisation de l'état de transition. Durant cette transition, les deux liaisons sigma sont formées à des vitesses différentes, ce qui peut générer des charges partielles dans l'état de transition qui peut être stabilisé par la distribution de la charge distribution dans les substituants conjuguées ;
- les dipolairophiles les plus polarisables sont les plus réactifs car les nuages électroniques diffus sont mieux adaptés pour initier le flux d'électrons ;
- les dipolairophiles avec une grande contrainte d'angle sont plus réactifs car cela accroit l'énergie de leur état fondamental ;
- l'augmentation de l'encombrement stérique de l'état de transition entrave la réactivité des réactifs et réduit drastiquement la vitesse de réaction ;
- les hétéro-dipolairophiles s’additionnent plus lentement, voire pas du tout, comparés aux diapolairophiles C=C à cause d'un gain d'énergie plus bas dans la transition entre une liaison pi et des liaisons sigma ;
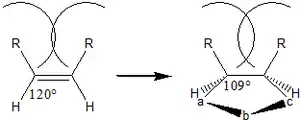
- l'isomérie du dipolairophile affecte la vitesse de réaction à cause de problèmes stériques. Les isomères trans sont plus réactifs (le trans-stilbène s'additionnera 27 fois plus rapidement sur le diphénylimine de nitrile) que le cis-stilbène) car durant la réaction, l'angle liaison de 120° se réduit à 109°, éclipsant les substituants du composé cis et accroissant l'encombrement stérique.
Orientation du produit
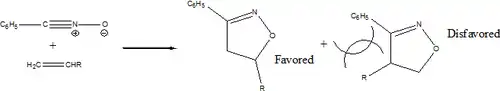
Si plusieurs règles générales ont été établies sur l'orientation du produit, la sélectivité réelle dans ces cycloadditions varie. Par exemple, dans une réaction de couplage azoture-alcyne sans catalyse par Cu(I), les produits 1,4 et 1,5 sont en ratio 1:1[4].
Dans les réactions avec des hétéro-dipolairophiles, l'orientation de l'addition est basée sur la maximisation des gains en énergie par la formation de liaison sigma durant la réaction. Cependant il existe plusieurs exceptions qui incluent des mécanismes réactionnels à étapes multiples.
Quand les dipolairophiles sont des alcènes ou des alcynes, la force principale dictant l'orientation de l'addition est les effets liés aux substituants, principalement stériques[1].
Classes de 1,3-dipôles
Huisgen et d'autres ont travaillé sur de nombreux 1,3-dipôles et dipolairophiles, incluant :
- les azotures et les alcynes réagissant par cycloaddition de Huisgen d'azoture-alcyne ;
- les composés diazo et les 1,3-dipôles réagissant par cycloaddition 1,3-dipolaire de diazoalcane ;
- les nitrones réagissant avec les alcènes pour former des isoxazolidines ;
- les oxydes de nitrile réagissant avec les alcynes pour former des isoxazoles ;
- les nitrilimines réagissant avec les alcynes pour former des pyrazoles ;
- l'ozonolyse qui débute par une cycloaddition 1,3-dipolaire de l'ozone ;
- les fullerènes et les nanotubes pouvant subir une cycloaddition 1,3-dipolaire avec l'ylure d'azométhine dans la réaction de Prato.
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « 1,3-Dipolar cycloaddition » (voir la liste des auteurs).
- (en) Rolf Huisgen, « Kinetics and Mechanism of 1,3-Dipolar Cycloadditions », Angewandte Chemie International Edition, vol. 2, no 11, , p. 633–645 (DOI 10.1002/anie.196306331, lire en ligne [abstract]).
- (en) Rolf Huisgen, « 1,3-Dipolar Cycloadditions. Past and Future », Angewandte Chemie International Edition, vol. 2, no 10, , p. 565–598 (DOI 10.1002/anie.196305651, lire en ligne [abstract]).
- (en) Huisgen, R., « The Concerted Nature of 1,3-Dipolar Cycoadditions and the Question of Diradical Intermediates », The Journal of Organic Chemistry, vol. 41, no 3, , p. 403–419 (DOI 10.1021/jo00865a001).
- (en) Vsevolod V. Rostovtsev, Luke G. Green, Valery V. Fokin et K. Barry Sharpless, « A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes », Angewandte Chemie International Edition, vol. 41, no 14, , p. 2596–22599 (DOI 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4).