Assemblage (bio-informatique)
En bio-informatique, l'assemblage consiste à aligner et/ou fusionner des fragments d'ADN ou d'ARN issus d'une plus longue séquence afin de reconstruire la séquence originale. Il s'agit d'une étape d'analyse in silico qui succède au séquençage de l'ADN ou de l'ARN d'un organisme unique, d'une colonie de clones (bactériens par exemple), ou encore d'un mélange complexe d'organismes.
Le problème de l'assemblage peut être comparé à celui de la reconstruction du texte d'un livre à partir de plusieurs copies de celui-ci, préalablement déchiquetées en petits morceaux.
Paradigmes d'assemblage
Les stratégies d'assemblage peuvent être organisées en 3 principaux paradigmes[1].
Glouton
Historiquement la première stratégie d'assemblage, celle-ci consiste à faire systématiquement le meilleur choix disponible sans possibilité de revenir sur ce choix plus tard. Le principal défaut de cette stratégie est qu'elle mène à des optimums locaux sans prendre en compte la relation globale entre les fragments. La plupart des assembleurs gloutons utilisent des heuristiques pour éviter le mauvais assemblage de séquences répétées. La plupart des premiers assembleurs tels que Phrap[2] ou TIGR[3] reposent sur ce paradigme, ainsi que quelques outils plus récents comme VCAKE[4].
"Overlap-Layout-Consensus" (OLC)
Cette stratégie d'assemblage se déroule en 3 étapes:
- Construction d'un graphe d'intervalles de chevauchement de fragments. Chaque fragment est un nœud du graphe, et une arête est créée entre deux fragments lorsque ceux-ci se chevauchent.
- Simplification du graphe. Des sous-graphes denses sont identifiés comme une collection de fragment qui se chevauchent entre eux et qui proviennent probablement de la même séquence originale.
- Extraction des séquences consensus. Une séquence consensus (contig) est générée à partir de l'ensemble des fragments de chaque sous-graphe.
Une variante de cette stratégie consiste à supprimer les liens transitifs du graphe de chevauchement pour construire un string graph.
Ce paradigme a notamment été rendu populaire par les travaux de Gene Myers intégrés dans l'assembleur Celera[5]. Les assembleurs de ce type ont dominé le monde de l'assemblage jusqu'à l'émergence des nouvelles technologies de séquençage (NGS). Ces dernières sont caractérisés par la production d'une très grande quantité de petits fragments (de quelques dizaines à plusieurs centaines de nucléotides), et les limites computationnelles de l'approche OLC ont rendu difficile l'application de cette stratégie aux données de séquençage moderne. Récemment, l'assembleur SGA[6] a introduit une nouvelle approche plus efficace basée sur des structures performantes pour l'indexation de chaines de caractère.
Graphe de De Bruijn
Les assembleurs basés sur un graphe de De Bruijn modélisent la relation entre des sous-chaines exactes extraites des fragments de séquençage. Dans un graphe de De Bruijn les nœuds sont des mots de taille k (k-mers), et les arêtes sont les chevauchements de taille k-1 entre les k-mers. Par exemple les 5-mers ACTAG et CTAGT partagent exactement 4 lettres. Les fragments ne sont pas directement modélisés dans ce paradigme, mais sont implicitement représentés par des chemins dans le graphe de De Bruijn.
Puisque les assembleurs basés sur ce paradigme reposent sur l'identification de chevauchements exacts, ceux-ci sont particulièrement sensibles à la présence d'erreurs de séquençage. Par conséquent, ces méthodes nécessitent l'utilisation d'étapes de correction des erreurs de séquençage avant et pendant l'assemblage afin d'obtenir des assemblages de haute qualité[1].
Cette approche a été popularisée par l'assembleur Euler, et a ensuite dominé le monde de l'assemblage des données modernes de séquençage à courts fragments, avec des outils comme Velvet[7], SOAPdenovo[8] et ALLPATHS[9].
Échafaudage du génome
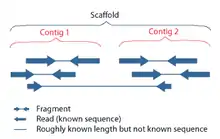
Un échafaudage, aussi appelé scaffold relie ensemble une série non contiguë de séquences génomiques. Il est ainsi constitué de séquences séparées par des lacunes (régions manquantes) de longueur connue. Les séquences qui sont liées sont généralement des séquences contiguës correspondant à des chevauchements de lectures.
Lors de la création d'une ébauche de génome, les lectures (appelées ‘reads’) individuelles d'ADN sont ensuite assemblées en contigs. Un contig est une longueur contiguë de lectures (reads) dont l'ordre des bases est connu avec un niveau de confiance élevé. Les contigs par la nature de leur assemblage, présentent des lacunes entre eux. Les lacunes se produisent lorsque les lectures des deux extrémités séquencées d'au moins un fragment chevauchent d'autres lectures dans deux contigs différents. Puisque les longueurs des fragments sont approximativement connues, le nombre de bases entre les contigs peut être estimé. L'étape suivante consiste à combler les lacunes entre ces contigs pour créer un échafaudage, ce qui peut être fait en utilisant la cartographie optique ou le séquençage par paires de matrices
Assemblage de-novo vs. avec référence
En termes de complexité et de temps requis, les assemblages de novo sont des ordres de grandeur plus lents et plus gourmands en mémoire que les assemblages de mappage. Cela est principalement dû au fait que l'algorithme d'assemblage doit comparer chaque lecture avec chaque autre lecture (une opération qui a une complexité temporelle naïve de O(n2)). Les assembleurs de génomes de novo actuels peuvent utiliser différents types d'algorithmes basés sur des graphes, tels que :
- L'approche Overlap/Layout/Consensus (OLC), qui était typique des assembleurs de données Sanger et repose sur un graphe de chevauchement.
- de Bruijn Graph (DBG), qui est la plus largement appliquée aux lectures courtes des plateformes Solexa et SOLiD. Il s'appuie sur des graphes K-mer, qui fonctionnent bien avec de grandes quantités de lectures courtes.
- gourmande basée sur les graphes, qui peut également utiliser l'une des approches OLC ou DBG. Avec des algorithmes gourmands basés sur des graphes, les contigs augmentent par extension gourmande, prenant toujours la lecture trouvée en suivant le chevauchement le plus élevés.
En se référant à la comparaison établie avec les livres déchiquetés dans l'introduction : alors que pour cartographier les assemblages, on aurait un livre très similaire comme modèle (peut-être avec les noms des personnages principaux et quelques emplacements modifiés), les assemblages de novo présentent un défi plus intimidant dans la mesure où l'on ne saurait d'avance si cela deviendrait un livre scientifique, un roman, un catalogue, voire plusieurs livres. De plus, chaque lambeau serait comparé à tous les autres lambeaux. La gestion des répétitions dans l'assemblage de novo nécessite la construction d'un graphe représentant les répétitions voisines. Une telle information peut être dérivée de la lecture d'un long fragment couvrant entièrement les répétitions ou seulement ses deux extrémités. D'un autre côté, dans un assemblage de mappage, les pièces avec plusieurs correspondances ou aucune correspondance sont généralement laissées à une autre technique d'assemblage à examiner.
Domaines d'application
Génomique
La Génomique est un domaine interdisciplinaire de la biologie (bio-informatique) axé sur la structure, la fonction, l'évolution, la cartographie et la modification des génomes.
Transcriptomique
La Transcriptomique est une domaine de la biologie qui étudier le Transcriptome d'un Organismes, c'est-à-dire la somme de toutes ses transcriptions d'ARN.
Protéomique
La protéomique est l'étude à grande échelle des protéomes.
Métagénomique
Le métagénome correspond à l’ensemble des génomes des micro-organismes présent dans un échantillon donné. La métagénomique est une méthodologie qui vise à étudier ce métagénome et donc le contenu génétique d'un échantillon issu d'un environnement complexe.
Métatranscriptomique
Le métatranscriptome correspond à l’ensemble des gènes exprimés par les micro-organismes présents dans un échantillon donné et à un instant donné. La métatranscriptomique est la méthodologie qui vise à étudier ce métatranscriptome.
Métaprotéomique
Le métaprotéome correspond à l’ensemble des protéines exprimées par les microorganismes présents dans un échantillon donné et à un instant donné. La métaprotéomique est la méthodologie qui vise à étudier ce métaprotéome.
Métabolomique
Le métabolome correspond à l’ensemble des métabolite détectés dans un échantillon. La métabolomique est la méthodologie qui vise à étudier ce métabolome.
Assembleurs disponibles
| Nom | Type | Technologies | Auteur | Publié / Dernière mise à jour | Licence* | Site |
|---|---|---|---|---|---|---|
| ABySS | (grands) génomes | Solexa, SOLiD | Simpson, J. et al. | 2008 / 2014 | NC-A | lien |
| ALLPATHS-LG | (grands) génomes | Solexa, SOLiD | Gnerre, S. et al. | 2011 | OS | lien |
| AMOS | génomes | Sanger, 454 | Salzberg, S. et al. | 2002? / 2011 | OS | lien |
| Arapan-M | génomes moyens (ex. E.coli) | All | Sahli, M. & Shibuya, T. | 2011 / 2012 | OS | lien |
| Arapan-S | (petits) génomes (virus et bactéries) | All | Sahli, M. & Shibuya, T. | 2011 / 2012 | OS | lien |
| Celera WGA Assembler / CABOG | (grands) génomes | Sanger, 454, Solexa | Myers, G. et al.; Miller G. et al. | 2004 / 2015 | OS | lien |
| CLC Genomics Workbench & CLC Assembly Cell | génomes | Sanger, 454, Solexa, SOLiD | CLC bio | 2008 / 2010 / 2014 | C | lien |
| Cortex | génomes | Solexa, SOLiD | Iqbal, Z. et al. | 2011 | OS | lien |
| DNA Baser Assembler | (petits) génomes | Sanger, 454 | Heracle BioSoft SRL | 06.2015 | C | lien |
| DNA Dragon | génomes | Illumina, SOLiD, Complete Genomics, 454, Sanger | SequentiX | 2011 | C | lien |
| DNAnexus | génomes | Illumina, SOLiD, Complete Genomics | DNAnexus | 2011 | C | lien |
| Edena | génomes | Illumina | D. Hernandez, P. François, L. Farinelli, M. Osteras, and J. Schrenzel. | 2008/2013 | OS | lien |
| Euler | génomes | Sanger, 454 (Solexa ?) | Pevzner, P. et al. | 2001 / 2006? | (C / NC-A?) | lien |
| Euler-sr | génomes | 454, Solexa | Chaisson, MJ. et al. | 2008 | NC-A | lien |
| Fermi | (grands) génomes | Illumina | Li, H. | 2012 | OS | lien |
| Forge | (grands) génomes, EST, metagénomes | 454, Solexa, SOLID, Sanger | Platt, DM, Evers, D. | 2010 | OS | lien |
| Geneious | génomes | Sanger, 454, Solexa, Ion Torrent, Complete Genomics, PacBio, Oxford Nanopore, Illumina | Biomatters Ltd | 2009 / 2013 | C | lien |
| Graph Constructor | (grands) génomes | Sanger, 454, Solexa, SOLiD | Convey Computer Corporation | 2011 | C | lien |
| IDBA (Iterative De Bruijn graph short read Assembler) | (grands) génomes | Sanger, 454,Solexa | Yu Peng, Henry C. M. Leung, Siu-Ming Yiu, Francis Y. L. Chin | 2010 | (C / NC-A?) | lien |
| LIGR Assembler (derived from TIGR Assembler) | génomes | Sanger | - | 2009/ 2012 | OS | lien |
| MaSuRCA (Maryland Super Read - Celera Assembler) | (grands) génomes | Sanger, Illumina, 454 | Aleksey Zimin, Guillaume Marçais, Daniela Puiu, Michael Roberts, Steven L. Salzberg, James A. Yorke | 2012 / 2013 | OS | lien |
| MIRA (Mimicking Intelligent Read Assembly) | génomes, ESTs | Sanger, 454, Solexa | Chevreux, B. | 1998 / 2014 | OS | lien |
| NextGENe | (petits génomes ?) | 454, Solexa, SOLiD | Softgenetics | 2008 | C | lien |
| Newbler | génomes, ESTs | 454, Sanger | 454/Roche | 2009/2012 | C | lien |
| PADENA | génomes | 454, Sanger | 454/Roche | 2010 | OS | lien |
| PASHA | (grands) génomes | Illumina | Liu, Schmidt, Maskell | 2011 | OS | lien |
| Phrap | génomes | Sanger, 454, Solexa | Green, P. | 1994 / 2008 | C / NC-A | lien |
| TIGR Assembler | génomes | Sanger | - | 1995 / 2003 | OS | « lien »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) |
| Ray[10] | génomes | Illumina, mix of Illumina and 454, paired or not | Sébastien Boisvert, François Laviolette & Jacques Corbeil. | 2010 | OS [GNU General Public License] | lien |
| Sequencher | génomes | traditional and next generation sequence data | Gene Codes Corporation | 1991 / 2009 / 2011 | C | lien |
| SeqMan NGen | (grands) génomes, exomes, transcriptomes, metagénomes, ESTs | Illumina, ABI SOLiD, Roche 454, Ion Torrent, Solexa, Sanger | DNASTAR | 2007 / 2014 | C | lien |
| SGA | (grands) génomes | Illumina, Sanger (Roche 454?, Ion Torrent?) | Simpson, J.T. et al. | 2011 / 2012 | OS | lien |
| SHARCGS | (petits) génomes | Solexa | Dohm et al. | 2007 / 2007 | OS | lien |
| SOPRA | génomes | Illumina, SOLiD, Sanger, 454 | Dayarian, A. et al. | 2010 / 2011 | OS | lien |
| SparseAssembler | (grands) génomes | Illumina, 454, Ion torrent | Ye, C. et al. | 2012 / 2012 | OS | lien |
| SSAKE | (petits) génomes | Solexa (SOLiD? Helicos?) | Warren, R. et al. | 2007 / 2014 | OS | lien |
| SOAPdenovo | génomes | Solexa | Li, R. et al. | 2009 / 2013 | OS | lien |
| SPAdes | (petits) génomes, single-cell | Illumina, Solexa, Sanger, 454, Ion Torrent, PacBio, Oxford Nanopore | Bankevich, A et al. | 2012 / 2015 | OS | lien |
| Staden gap4 package | BACs (, petits génomes ?) | Sanger | Staden et al. | 1991 / 2008 | OS | lien |
| Taipan | (petits) génomes | Illumina | Schmidt, B. et al. | 2009 / 2009 | OS | lien |
| VCAKE | (petits) génomes | Solexa (SOLiD?, Helicos?) | Jeck, W. et al. | 2007 / 2009 | OS | lien |
| Phusion assembler | (grands) génomes | Sanger | Mullikin JC, et al. | 2003 / 2006 | OS | lien |
| Quality Value Guided SRA (QSRA) | génomes | Sanger, Solexa | Bryant DW, et al. | 2009 / 2009 | OS | lien |
| Velvet | (petits) génomes | Sanger, 454, Solexa, SOLiD | Zerbino, D. et al. | 2007 / 2011 | OS | lien |
| Canu[11] | génomes | PacBio, Oxford Nanopore | Koren, S. et al. | 2017 / 2018 | OS | lien |
| *Licences: OS = Open Source; C = Commercial; C / NC-A = Commercial mais gratuit pour non-commercial et académiques; Parenthèses = probablement C / NC-A | ||||||
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Sequence assembly » (voir la liste des auteurs).
- (en) Niranjan Nagarajan et Mihai Pop, « Sequence assembly demystified », Nature Reviews Genetics, vol. 14, , p. 157–167 (ISSN 1471-0056, DOI 10.1038/nrg3367, lire en ligne, consulté le )
- « Phred, Phrap, and Consed », sur www.phrap.org (consulté le )
- « JCVI: Abstract », sur www.jcvi.org (consulté le )
- « VCAKE », sur SourceForge (consulté le )
- « wgs-assembler », sur wgs-assembler.sourceforge.net (consulté le )
- « jts/sga », sur GitHub (consulté le )
- « Velvet: a sequence assembler for very short reads », sur www.ebi.ac.uk (consulté le )
- BGI,Bioinformatics Center,SOAP Team,GentleYang, « SOAP :: Short Oligonucleotide Analysis Package », sur soap.genomics.org.cn (consulté le )
- « ALLPATHS-LG | High quality genome assembly from low cost data », sur www.broadinstitute.org (consulté le )
- (en) Sébastien Boisvert, François Laviolette et Jacques Corbeil, « Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies », Journal of Computational Biology, vol. 17, no 11, , p. 1519–33 (PMID 20958248, PMCID 3119603, DOI 10.1089/cmb.2009.0238)
- (en) Sergey Koren, Brian P. Walenz, Konstantin Berlin et Jason R. Miller, « Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation », Genome Research, vol. 27, no 5, , p. 722–736 (ISSN 1088-9051 et 1549-5469, PMID 28298431, DOI 10.1101/gr.215087.116, lire en ligne, consulté le )