Activation de liaison C-H
L'activation de liaison carbone-hydrogène, ou activation C-H, est une réaction qui rompt une liaison carbone-hydrogène[1] - [2] - [3] - [4] - [5] - [6] - [7] - [8] - [9] - [10]. L'expression se limite le plus souvent à la désignation de réaction mettant en jeu des complexes organométalliques et qui procèdent par la coordination d'un hydrocarbure à la sphère interne du métal, soit par un intermédiaire "alcane ou aryle complexe" soit comme un état de transition menant à un intermédiaire "M-C" [11] - [12] - [13]. Cette définition inclut aussi le fait que, lors de la rupture de la liaison C-H, l'espèce hydrocarbure reste associée à la sphère interne et sous l'influence du métal.
Les études théoriques aussi bien que les investigations expérimentales indiquent que la liaison C-H, usuellement considérée comme non réactive, peut être rompue par coordination. De nombreux efforts ont été effectués pour le design et la synthèse de nouveaux réactifs et catalyseurs dans ce domaine. Un des principaux intérêts au développement de cette recherche, est l'idée que l'activation C-H pourrait permettre la conversion d'alcanes peu onéreux et disponibles en grandes quantités en des composés fonctionnalisés de plus haute valeur ajoutée.
Historique
La première réaction d'activation C-H est souvent attribuée à Otto Dimroth qui, en 1902, rapporta la réaction du benzène avec l'acétate de mercure(II) mais certains académiques ne considèrent pas cette réaction comme étant réellement de l'activation C-H. Comme l'ont observé Goldman & Goldberg[11], l'activation C-H a certains aspects de l'activation H-H, puisque les deux peuvent être effectuées par oxydation électrophile ou nucléophile.
La première véritable activation C-H a été rapportée par Joseph Chatt[14] en 1965 avec l'insertion d'un atome de ruthénium, ligandé par du dmpe, dans une liaison C-H du naphtalène. En 1969, A.E. Shilov a publié l'échange isotopique entre du méthane et de l'eau lourde en présence de tetrachloroplatinate de potassium. Le mécanisme proposé faisait intervenir une liaison méthane-Pt(II). En 1972, le groupe de Shilov a pu produire du méthanol et du chlorométhane à partir de méthane et d'eau, en présence de tetrachloroplatinate de potassium et d'une quantité catalytique d'hexachloroplatinate de potassium. Ces travaux furent cependant longtemps ignorés de la communauté scientifique, Shilov ayant publié sous l'ère de l'Union Soviétique, pendant la guerre froide. Ce "système" de Shilov est aujourd'hui considéré comme le premier véritable exemple de fonctionnalisation d'alcane par voie catalytique[11].
De leur côté, M.L.H. Green[15] rapporta en 1970 l'insertion de tungstène (sous forme de Cp2WH2) dans les liaisons C-H du benzène par activation photochimique et M. Whitesides[16] a été le premier en 1979 a mener une activation C-H intramoléculaire sur un composé aliphatique.
L'avancée majeure suivante fut effectuée de manière indépendante par deux groupes en 1982. R.G. Bergman démontra l'activation photochimique des liaisons C-H du cyclohexane et du néopentane pour former le complexe métallique Cp*Ir(PMe3)H(C6H5)[17] tandis que W.A.G. Graham découvrit que les mêmes hydrocarbures réagissent avec Cp*Ir(CO)2 pour former un complexe d'iridium[18].
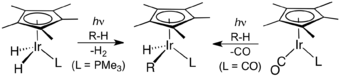 Activation C-H par Bergman et al. (à gauche) et Graham et al.
Activation C-H par Bergman et al. (à gauche) et Graham et al.
Plus tard, en 1999 et 2000, Hartwig développa la borylation régiosélective d'aryles et alcanes par un complexe de rhodium. Dans le cas des alcanes, la formation exclusive du dérivé boré terminal a été observée[19].
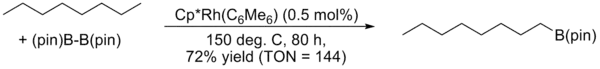 Hartwig Borylation 2000
Hartwig Borylation 2000
Développement
Dans une étude, le pentane est sélectivement converti en le dérivé halogéné 1-iodopentane, à l'aide d'un complexe de tungstène[20].
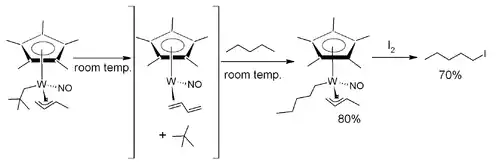 Activation C-H du pentane
Activation C-H du pentane
Le complexe de tungstène est formé d'une unité Cp*, d'un groupement nitroso, d'une unité butène et d'un ligand néopentane. Ce complexe est thermodynamiquement instable et, au moment de la dissolution dans le pentane, perd son ligand néopentane et se coordonne au pentane. Ce changement de ligand s'opère via le complexe à 16 électrons possédant un ligand butadiène formé par élimination bêta. Lors d'une seconde étape, l'iode est ajoutée à −60 °C et on observe la formation de 1-iodopentane.
Les liaisons C-H aromatiques peuvent aussi être activées par des complexes métalliques, malgré le fait qu'elles soient relativement peu réactives. Un exemple est le couplage d'oléfines de Murai. Dans cette réaction, un complexe de ruthénium réagit avec la N,N-diméthylbenzylamine dans un processus de cyclométallation comprenant une activation C-H.
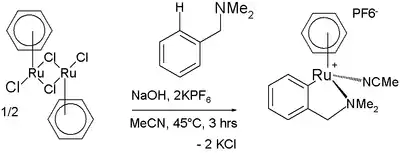 Cyclométallation d'une benzylamine substituée
Cyclométallation d'une benzylamine substituée
L'activation d'une liaison C-H vinylique est également envisageable, comme l'a montré Yotphan en 2008 par la synthèse d'une amine bicyclique en présence d'un complexe de rhodium[21].
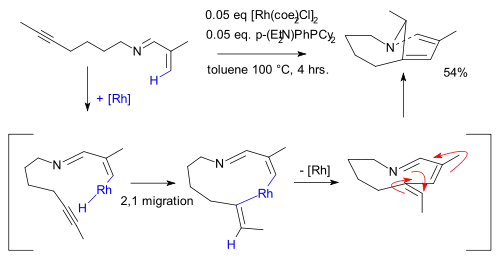 Activation C-H par Yotphan, 2008
Activation C-H par Yotphan, 2008
Il a été également démontré par R.A. Periana et al. que les sels et complexes de certains métaux de transitions tels que Pt, Pd, Au et Hg peuvent réagir avec le méthane dans l'acide sulfurique pur former de l'acide méthylsulfurique, avec des rendements variés selon le métal, et de hautes sélectivités[22] - [23].
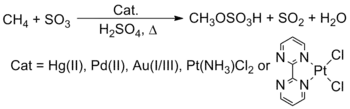
Conditions Réactionnelles
La plupart des réactions d'activation C-H nécessitent des conditions plutôt dures (haute température, milieux fortement acides ou basiques, oxydants forts), ce qui diminue beaucoup leur attractivité. Toutefois, de plus en plus de réactions aux conditions douces ont été développées, étendant significativement le champ d'application de ces transformations[24]. L'organocatalyse a été récemment utilisée comme approche sans métal et peu onéreuse[25].
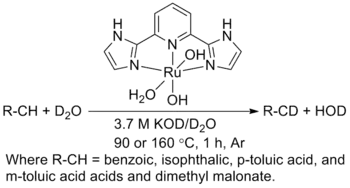
Récemment, Hashigushi et al. ont découvert que les hydroxydes métalliques et les aqua-complexes pouvaient être utilisés pour mener des réactions d'activation C-H sur des substrats solubles dans l'eau sous conditions fortement basiques[26]. Ils ont notamment montré qu'un précatalyseur au ruthénium avec une pince NNN (2,6-diimidizopyridine, IPI), une fois dissout en milieu aqueux basique, générait le complexe mixte correspondant, Ru(IPI(OH)n(H2O)m). Ce complexe s'est ensuite montré capable de catalyser l'échange H/D entre l'aryle et la base, avec un accroissement de la concentration en KOH en fonction de la réduction du Ru(III) en Ru(II). Ce fut la première démonstration d'une activation C-H accélérée par une base.
Voir aussi
- Couplage oxydant du méthane (en)
Références
- “Alkane C−H activation and functionalization with homogeneous transition metal catalysts: a century of progress – a new millennium in prospect”, R. H. Crabtree, J. Chem. Soc., Dalton Trans. 2001, 17, 2437–2450. DOI 10.1039/B103147N
- "Designing Catalysts for Functionalization of Unactivated C–H Bonds Based on the CH Activation Reaction" B. G. Hashiguchi, S. M. Bischof, M. M. Konnick, R. A. Periana. Accounts of Chemical Research 2012; ASAP. DOI 10.1021/ar200250r
- “Organometallic alkane CH activation”, R. H. Crabtree, J. Organometal. Chem. 2004, 689, 4083–4091. DOI 10.1016/j.jorganchem.2004.07.034
- “Mechanistic Aspects of C−H Activation by Pt Complexes”, M. Lersch, M.Tilset, Chem. Rev. 2005, 105, 2471−2526. DOI 10.1021/cr030710y
- “Recent Advances in the Platinum-mediated CH Bond Functionalization”, A. N. Vedernikov, Curr. Org. Chem. 2007, 11, 1401−1416.
- “Catalytic C−H functionalization by metalcarbenoid and nitrenoid insertion”, H. M. L. Davies, J. R. Manning, Nature, 2008, 451, 417−424, DOI 10.1038/nature06485
- "Mechanisms of C−H bond activation: rich synergy between computation and experiment”, Y. Boutadla, D. L. Davies, S. A. Macgregor, A. I. Poblador-Bahamonde, Dalton Trans. 2009, 5820−5831. DOI 10.1039/B904967C
- “C−H Bond Activation in Transition Metal Species from a Computational Perspective”, D. Balcells, E. Clot, O. Eisenstein, Chem. Rev. 2010, 110, 749–823. DOI 10.1021/cr900315k
- “Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions”, T. W. Lyons, M. S. Sanford, Chem. Rev. 2010, 110, 1147–1169. DOI 10.1021/cr900184e
- “Selectivity enhancement in functionalization of C−H bonds: A review”, G. B. Shul’pin, Org. Biomol. Chem. 2010, 8, 4217–4228. DOI 10.1039/c004223d
- Organometallic C−H Bond Activation: An Introduction Alan S. Goldman and Karen I. Goldberg ACS Symposium Series 885, Activation and Functionalization of C−H Bonds, 2004, 1–43
- Arndtsen, B. A.; Bergman, R. G.; Mobley, T. A.; Peterson, T. H. “Selective Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution.” Accounts of Chemical Research, 1995: 28 (3) 154–162.
- Periana, R. A.; Bhalla, G.; Tenn, W. J., III, Young, K. J. H.; Liu, X. Y.; Mironov, O.; Jones, C.; Ziatdinov, V. R. “Perspectives on some challenges and approaches for developing the next generation of selective, low temperature, oxidation catalysts for alkane hydroxylation based on the C−H activation reaction.” Journal of Molecular Catalysis A: Chemical, 2004: 220 (1) 7–25. DOI 10.1016/j.molcata.2004.05.036
- The tautomerism of arene and ditertiary phosphine complexes of ruthenium(0), and the preparation of new types of hydrido-complexes of ruthenium(II) J. Chatt and J. M. Davidson, J. Chem. Soc. 1965, 843 DOI 10.1039/JR9650000843
- Formation of a tangsten phenyl hydride derivatives from benzene M. L. Green, P. J. Knowles, J. Chem. Soc. D, 1970, (24), 1677–1677 DOI 10.1039/C29700001677
- Thermal generation of bis(triethylphosphine)-3,3-dimethylplatinacyclobutane from dineopentylbis(triethylphosphine)platinum(II) Paul Foley, George G.M. Whitesides J. Am. Chem. Soc. 1979; 101(10); 2732–2733. DOI 10.1021/ja00504a041
- Carbon–hydrogen activation in saturated hydrocarbons: direct observation of M + R−H → M(R)(H) Andrew H. Janowicz, Robert G. Bergman J. Am. Chem. Soc.; 1982; 104(1); 352–354.DOI 10.1021/ja00365a091
- Oxidative addition of the carbon–hydrogen bonds of neopentane and cyclohexane to a photochemically generated iridium(I) complex James K. Hoyano, William A. G. Graham J. Am. Chem. Soc. 1982; 104(13); 3723–3725. DOI 10.1021/ja00377a032
- Thermal, Catalytic, Regiospecific Functionalization of Alkanes Huiyuan Chen, Sabine Schlecht, Thomas C. Semple, John F. Hartwig Science 2000; 287(5460); 1995–1997. DOI 10.1126/science.287.5460.1995
- Selective Activation and Functionalization of Linear Alkanes Initiated under Ambient Conditions by a Tungsten Allyl Nitrosyl Complex Jenkins Y. K. Tsang, Miriam S. A. Buschhaus, and Peter Legzdins J. Am. Chem. Soc.; 2007; 129(17) p. 5372–5373; (Communication) DOI 10.1021/ja0713633
- The Stereoselective Formation of Bicyclic Enamines with Bridgehead Unsaturation via Tandem C−H Bond Activation/Alkenylation/ Electrocyclization Sirilata Yotphan, Robert G. Bergman, and Jonathan A. Ellman J. AM. CHEM. SOC. 2008, 130, 2452–2453 DOI 10.1021/ja710981b
- (en) R.A. Periana, D.J. Taube, E.R. Evitt, D.G. Loffler, P.R. Wentrcek, G. Voss et T. Masuda, « A Mercury-Catalyzed, High-Yield System for the Oxidation of Methane to Methanol », Science, vol. 259, no 5093, , p. 340–343 (PMID 17832346, DOI 10.1126/science.280.5363.493f)
- (en) R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh et H. Fujii, « Platinum Catalysts for the High-Yield Oxidation of Methane to a Methanol Derivative », Science, vol. 280, no 5363, , p. 560–564 (PMID 9554841, DOI 10.1126/science.280.5363.560, Bibcode 1998Sci...280..560P)
- Towards Mild Metal-Catalyzed C-H Bond Activation J. Wencel-Delord, T. Dröge, F. Liu, F. Glorius Chem. Soc. Rev. 2011, 40, 4740-4761 DOI 10.1039/C1CS15083A
- Pan, S. C. “Organocatalytic C–H activation reactions” Beilstein J. Org. Chem. 2012, 8, 1374–1384. DOI 10.3762/bjoc.8.159 (Open Access)
- Acceleration of Nucleophilic CH Activation by Strongly Basic Solvents B. G. Hashiguchi, K. J. H. Young, Muhammed Yousufuddin, W. A. Goddard, III, R. A. Periana J. Am. Chem. Soc. 2010, 132, 12542-12545 DOI 10.1021/ja102518m
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Carbon–hydrogen bond activation » (voir la liste des auteurs).