Neurofibromatose
Les neurofibromatoses (NF) sont des maladies génétiques, ce sont des maladies orphelines. Elles font partie des phacomatoses dont le mécanisme est un problème de différenciation du tissu ectodermique chez l'embryon.
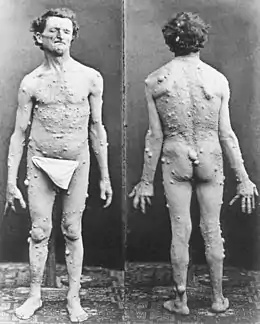
| Spécialité | Génétique médicale et neurologie |
|---|
| CISP-2 | A90 |
|---|---|
| CIM-10 | Q85.0 |
| CIM-9 | 237.7 |
| ICD-O | 9540/0 |
| OMIM | 162200 101000 |
| eMedicine | 1112001 |
| MeSH | D017253 |
| Patient UK | Neurofibromatosis |
![]() Mise en garde médicale
Mise en garde médicale
Il existe plusieurs types de neurofibromatoses qui n'ont aucun rapport entre elles:
- La neurofibromatose de type I (NF1 - maladie de Recklinghausen - 1881, 1988)
- La neurofibromatose de type II (NF2 - tumeurs bénignes : schwannomes vestibulaires bilatéraux - 1822, 1920, 1993)
- La neurofibromatose de type III, ou schwannomatose (NF3 - tumeurs bénignes : schwannomes non vestibulaires bilatéraux )
La neurofibromatose de type 1
La neurofibromatose de type 1 (NF1) est une maladie génétique de transmission autosomique dominante, liée à une mutation du gène NF1 codant la neurofibromine.
La génétique des neurofibromatoses est reprise dans un chapitre dédié. Sa prévalence est de 1/40001–4 et son incidence estimée à une naissance sur 30001,2,5–7.
Son expression phénotypique est variable même entre les membres d’une famille. Sa pénétrance est complète à l’âge de 8 ans : 97% des patients atteints de NF1 remplissent les critères diagnostiques à 8 ans
La neurofibromatose de type 2
La neurofibromatose de type 2 (NF2) est une maladie génétique rare. Elle se caractérise par le développement quasi systématique de schwannomes vestibulaires (ou neurinomes de l’acoustique), c’est-à-dire de tumeurs situées le long des nerfs auditifs pouvant entraîner des troubles de l’audition.
S’y associent aussi fréquemment des méningiomes, tumeurs bénignes de la méninge (enveloppe du cerveau). Il peut s’y associer aussi des tumeurs bénignes comprimant la moelle épinière et pouvant entraîner des troubles de la marche ou des faiblesses des bras ou des jambes.
Les symptômes sont variables d’un sujet à l’autre y compris à l’intérieur d’une même famille allant de formes mineures à des formes plus sévères. L’évolution des tumeurs et des symptômes n’est pas prévisible et donc l’éventuel traitement doit être adapté au cas par cas. La surdité peut être améliorée par des appareillages sophistiqués nécessitant parfois une prise en charge par des équipes spécialisées.
Les schwannomatoses
Les schwannomatoses constituent des syndromes génétiques de prédisposition à développer des schwannomes multiples, et plus rarement des méningiomes1. Ce sont des maladies rares, les données épidémiologiques les plus récentes faisant état d’une prévalence de 1 pour 126 315 personnes et d’une incidence de 1 pour 68 956 naissances2.
Contrairement aux patients porteurs de neurofibromatoses, les patients porteurs de schwannomatoses ont souvent des symptômes non spécifiques expliquant le délai de plusieurs années entre l’apparition des premiers symptômes et le diagnostic. Ce retard est accentué par le fait que les formes familiales, dans lesquelles au moins un des deux parents est atteint, sont beaucoup plus rares que les formes sporadiques, dans lesquelles aucun des deux parents n’est atteint.
Au sein même de chacune des deux formes cliniques (familiale ou sporadique), il existe un sous-groupe de patients présentant des schwannomatoses dites segmentaires dans lesquelles un seul membre est atteint.
Notes et références
CEntre de REférence des Neurofibromatoses France, CERENEF,
La neurofibromatose de type 1 – CERENEF (centre-neurofibromatoses.fr)