FASTQ
Le format FASTQ est un format de fichier texte permettant de stocker à la fois des séquences biologiques (uniquement des séquences nucléiques) et les scores de qualité associés. La séquence et le score sont chacune codées avec un seul caractère ASCII. Ce format a été initialement développé par le Wellcome Trust Sanger Institute afin de lier un fichier de séquence au format FASTA aux données de qualité correspondantes mais est récemment devenu le standard de facto pour le stockage des sorties de séquenceurs à haut débit[1].
Historique
Le format FASTQ fut inventé par Jim Mullikin au Wellcome Trust Sanger Institute vers la fin du XXe siècle[1]. À cette époque, les projets de séquençage commençaient à prendre des envergures considérables donnant naissance à des projets tel que le Projet Génome Humain. Ces projets générèrent des quantités de séquences de plus en plus grandes, nécessitant un traitement automatisé. Ce besoin d'automatisation aboutit entre autres à la création du programme Phred permettant d'attribuer aux bases séquencées des scores de qualité. Ainsi pour chaque séquence générée, deux fichiers étaient créés : un fichier FASTA contenant la séquence nucléique et un fichier QUAL contenant sous format numérique les scores associés à chacune des bases de la séquence[1]. Afin de ne manipuler qu'un unique fichier, les formats FASTA et QUAL furent fusionnés en un seul format, le format FASTQ. Cependant, bien que ce nouveau format fut largement diffusé et adopté, aucune spécification officielle ne fut donnée[1].
Au début du XXIe siècle, la multiplication des projets de séquençage à travers le monde aboutit à l'émergence de nouvelles techniques de séquençage qui se répandirent rapidement. Ces nouvelles techniques étant elles aussi basées sur une automatisation poussée du séquençage, elles adoptèrent naturellement le format FASTQ. Néanmoins, comme aucune spécification officielle n'encadrait ce format, quelques variants incompatibles entre eux et avec le format original virent le jour[1]. Toutefois, un effort de spécification de ce format a été entrepris par la communauté scientifique et particulière par l'Open Bioinformatics Foundation[1].
Format
Description
Un fichier FASTQ utilise en principe 4 lignes par séquence[1]. La ligne 1 commence par un caractère "@" suivi de l'identifiant de la séquence et éventuellement d'une description (de la même façon qu'un fichier au format FASTA, le "@" remplaçant ici le ">"). La ligne 2 contient la séquence nucléique brute. La ligne 3 commence par un caractère "+", parfois suivi par la répétition de l'identifiant de la séquence et de sa description si celle-ci est présente. La ligne 4 contient les scores de qualité associés à chacune des bases de la séquence de la ligne 2 et doit avoir exactement le même nombre de symboles que la ligne 2.
Codage du score de qualité
À l'origine, les scores de qualité phred des fichiers QUAL étaient codés sous format numérique, chaque nombre étant séparé par un espace et composé d'un ou deux chiffres[1]. Ce système ne peut être appliqué au format FASTQ car pour chaque base codée par un unique caractère (A, C, G ou T) doit correspondre un score devant être lui aussi codé par un unique caractère. L'utilisation du code ASCII permet de surmonter cette contrainte en faisant correspondre, à un caractère, son code en base dix, ce code correspondant alors au score de qualité. Toutefois, les 32 premiers caractères du code ASCII étant des caractères de contrôle (ne codant pas pour un caractère latin et donc ne pouvant être lu par un humain) et le 32e caractère correspondant à l'espace difficilement interprétable, il a été décidé que le premier code utilisé pour coder le score de qualité 0 serait 33 (correspondant au caractère "!")[1]. D'autre part, le code ASCII ne possédant que 128 caractères, le 128e étant un caractère de contrôle (code 127), ce système de codage possède donc une limite supérieure correspondant à un score de 93 (126-33). Par ailleurs, le décalage entre la valeur du score phred et le code ASCII correspondant est généralement symbolisé par la notation Phred+33.
Exemple type
Voici la présentation d'un fichier FASTQ minimal :
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
La version originale de ce format (nommée fastq-sanger[1]) autorisait le retour à la ligne dans la séquence et les scores de qualité, mais ce comportement est peu recommandé car il complique la tâche de lecture du fichier (ou parsing) en raison du choix malavisé du "@" et du "+", ces caractères pouvant en effet coder des scores de qualité.
Extension de fichier
Il n'existe pas à l'heure actuelle une extension de fichier standard pour les fichiers FASTQ mais les extensions .fastq et .fq sont les plus fréquemment utilisées.
Variants de format
Le format FASTQ n'ayant eu aucune spécification officielle, des variations plus ou moins importantes sont observées dans les fichiers créés par les différentes plates-formes de séquençage. Ainsi trois variants de format sont reconnus officiellement par l’Open Bioinformatics Foundation : le variant Sanger fastq-sanger, le variant Solexa/Illumuna fastq-solexa et le variant Illumina fastq-illumina[1]. L'harmonisation du format initiée par l'Open Bioinformatics Foundation utilise comme base le variant original fastq-sanger.
Ces variations concernent principalement le formatage de l'identifiant associé à chaque séquence, la méthode de calcul des scores de qualité ainsi que leur codage. Ces deux derniers points ont rendu les formats incompatibles entre eux, nécessitant des conversions de fichiers.
Identifiants de séquences Illumina
Bien que l'identifiant n'ait reçu aucune spécification tant sur le nombre de caractères que leur nature et sur l'information minimale qu'il doit contenir, des normes propres à chaque plate-forme ont émergé. Concernant les plates-formes Illumina, l'identifiant originellement utilisé fut celui créé par Solexa avant de subir quelques variations lors de développements ultérieurs.
Les séquences issues des plates-formes Illumina étaient identifiées de la manière suivante jusqu'à la version 1.4 du pipeline du Genome Analyzer d'Illumina :
@HWUSI-EAS100R:6:73:941:1973#0/1
| HWUSI-EAS100R | Nom unique de l'instrument |
|---|---|
| 6 | numéro de piste de la lame (flowcell lane) |
| 73 | numéro de zone (tile) au sein de la piste |
| 941 | [[Repérage dans le plan et dans l'espace#Repérage dans le plan|coordonnée x]] du cluster au sein de la zone |
| 1973 | [[Repérage dans le plan et dans l'espace#Repérage dans le plan|coordonnée y]] du cluster au sein de la zone |
| #0 | numéro d'index pour les échantillons multiplexés (si aucun multiplex, 0) |
| /1 | le membre de la paire, /1 ou /2 (uniquement en cas de séquences appariées (paired-end ou mate-pair reads)) |
Depuis la version 1.4, le pipeline remplace le numéro d'index #0 par la séquence de l'index (étiquette nucléotidique ou tag) NNNNNN afin d'identifier les échantillons multiplexés.
Avec la version 1.8 de CASAVA, le format de l'identifiant a de nouveau changé :
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
| EAS139 | identifiant unique de l'instrument |
|---|---|
| 136 | identifiant du projet (run) |
| FC706VJ | identifiant de la lame (flowcell) |
| 2 | numéro de la piste de la lame (flowcell lane) |
| 2104 | numéro de la zone (tile) au sein de la piste |
| 15343 | [[Repérage dans le plan et dans l'espace#Repérage dans le plan|coordonnée x]] du cluster au sein de la zone |
| 197393 | [[Repérage dans le plan et dans l'espace#Repérage dans le plan|coordonnée y]] du cluster au sein de la zone |
| 1 | membre de la paire, 1 ou 2 (uniquement en cas de séquences appariées (paired-end ou mate-pair reads)) |
| Y | indication de passage du filtre, Y (pour yes) indique une mauvaise séquence (read), sinon N (pour no) |
| 18 | 0 lorsque aucun des bit contrôles n'est activé, sinon c'est un nombre |
| ATCACG | index (étiquette nucléotidique ou tag) de la séquence |
Archives de séquençage
Les fichiers FASTQ déposés aux Archives de séquençage (Sequence Read Archive) du NCBI, de l'EBI ou de la DDBJ ont un identifiant et une description comme suit :
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC +SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
Dans cet exemple, l'identifiant original est remplacé par un identifiant assigné par les archives contenant une référence unique au sein des Archives de séquençage suivi d'une description contenant l'identifiant original produit lors du séquençage (comme décrit précédemment) ainsi que la longueur de la séquence.
Il est à noter que les archives convertissent systématiquement les données FASTQ codées à l'aide des variants Solexa/Illumina vers le variant Sanger (voir la section codage du score de qualité).
Calcul du score de qualité
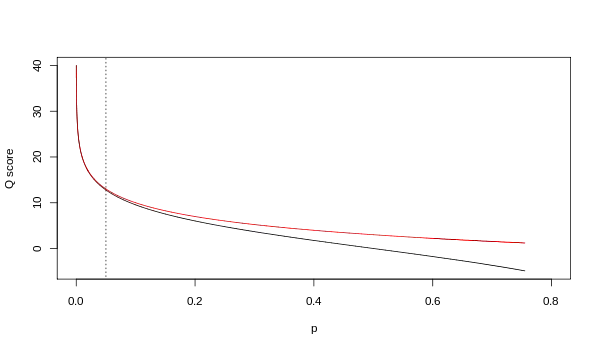
Un score de qualité Q est un entier relié de façon logarithmique à une probabilité d'erreur p. Cette probabilité d'erreur est calculée lors de l'identification d'une base et correspond à la probabilité qu'il s'agisse d'une mauvaise identification.
Deux équations différentes ont été utilisées pour calculer ce score de qualité. La première est le standard de Sanger pour déterminer la fidélité dans l'identification des bases, aussi connu comme le score de qualité phred :

Le pipeline Solexa/Illumina utilisait initialement une équation différente, calculant le logarithme de p/(1-p) au lieu de celui de la probabilité p :

Bien que les valeurs de score soient identiques au niveau de l'asymptote verticale correspondant aux scores les plus élevés, elles diffèrent concernant les scores les plus faibles (c'est-à-dire pour des probabilités p > 0.05 correspondant à des scores Q < 13)
Cependant, il existe une certaine ambigüité sur la méthode de calcul utilisée en réalité par Illumina. Le guide de l'utilisateur de la version 1.4 du pipeline[2] précise que « Les scores sont définis selon Q=10*log10(p/(1-p)) [sic], où p est la probabilité que la base identifiée corresponde à la base en question » (Appendix B, page 122). Avec le recul, il semblerait que cette définition aurait été erronée. Le guide de l'utilisateur de la version 1.5 du pipeline[3] énonce cette nouvelle description : « Les changements importants dans le Pipeline v1.3 [sic]. La méthode de calcul du score de qualité à changer pour celle de la méthode Phred (c'est-à-dire Sanger), chaque score étant codé avec un caractère ASCII correspondant à la valeur Phred auquel aura été ajoutée 64. Le score Phred d'une base est calculé comme suit : =-10 (e), où e est la probabilité estimée qu'une base soit erronée » (What's New, page 5).


Codage du score de qualité
- Le variant Sanger code les scores de qualité phred de 0 à 93 en utilisant les codes ASCII 33 à 126 (Phred+33). Bien qu'à partir des données de séquences brutes les scores de qualité dépassent rarement 60, des scores plus élevés sont possibles au sein des assemblages ou des alignements de séquences. Ce codage est également utilisé dans le format de fichier SAM[4]. Produit à la fin février 2011, la nouvelle version 1.8 du pipeline Illumina code directement les fichiers FASTQ dans le variant Sanger comme il a été annoncée sur le forum Seqanswers.com[5].
- Le variant Solexa/Illumina 1.0 code les scores de qualité Solexa/Illumina de -5 à 62 utilisant les codes ASCII 59 à 126 (Phred+64). Cependant, les scores Solexa des données de séquences brutes sont prévues uniquement entre -5 et 40.
- À partir de la version 1.3 et jusqu'à la version 1.8 du pipeline Illumina, le format utilisé code les scores de qualité phred de 0 à 62 utilisant les codes ASCII 64 à 126 (Phred+64). Cependant, les scores de qualité des données de séquences brutes sont prévues uniquement entre 0 et 40.
- À partir de la version 1.5 et jusqu'à la version 1.8 du pipeline Illumina, les scores de qualité phred allant de 0 à 2 ont une signification légèrement différente de ceux des autres systèmes de codage utilisés par le pipeline. Les valeurs 0 et 1 ne sont plus utilisées et la valeur 2, codée par le code ASCII 66 correspondant au caractère "B", est utilisée aussi à la fin des séquences comme indicateur de contrôle qualité de segment de séquence (Read Segment Quality Control Indicator)[6]. Le manuel Illumina[7] indique ceci en page 30 : « Si des fins de séquences avec une importante portion de basse qualité (Q15 ou inférieur), tous les scores de qualité dans cette portion sont remplacés par la valeur 2 (codée par la lettre B dans le système de codage texte des scores de qualité d'Illumina). (…) Cet indicateur Q2 ne prédit pas un taux d'erreur spécifique mais indique plutôt qu'une portion terminale spécifique d'une séquence ne devrait pas être utilisée dans les analyses futures ». Ce score de qualité codé par la lettre "B" peut aussi apparaître ailleurs dans la séquence au moins jusqu'à la version 1.6, comme dans cet exemple :
@HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT +HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 efcfffffcfeefffcffffffddf`feed]`]_Ba_^__[YBBBBBBBBBBRTT\]][]dddd`ddd^dddadd^BBBBBBBBBBBBBBBBBBBBBBBB
Une autre interprétation de ce codage ASCII a été proposée[8]. Ainsi, dans les projets Illumina utilisant les contrôles PhiX, le caractère "B" était assimilé à un "score de qualité inconnu". Le score "B" était attribué grossièrement lorsque le score de qualité était inférieur de 3 points au score moyen observé lors d'un séquençage donné.
- À partir de la version 1.8 du pipeline Illumina, le codage des scores de qualité a de nouveau suivi celui utilisé par le variant Sanger(Phred+33).
Pour les séquences brutes, l'intervalle de scores dépendra de la technique de séquençage et du programme d'identification de bases utilisés mais ne pourra dépasser typiquement une valeur de 40. Récemment, l'amélioration par Illumina de sa technique conduisit à l'obtention de scores de 41, ce qui a engendré des bogues dans de nombreux scritps et programmes n'étant pas prévu pour interpréter un score au-delà de 40. Pour des séquences alignées et des séquences consensus, des scores plus élevés sont fréquents.
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS.....................................................
..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX......................
...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII......................
.................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ......................
LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL....................................................
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
| | | | | |
33 59 64 73 104 126
0........................26...31.......40
-5....0........9.............................40
0........9.............................40
3.....9.............................40
0........................26...31........41
S - Sanger Phred+33, scores des séquences brutes compris entre 0 et 40
X - Solexa Phred+64, scores des séquences brutes compris entre -5 et 40
I - Illumina 1.3+ Phred+64, scores des séquences brutes compris entre 0 et 40
J - Illumina 1.5+ Phred+64, scores des séquences brutes compris entre 3 et 40
avec 0=inutilisé, 1=inutilisé, 2=Indicateur de contrôle qualité de segment de séquence (en gras)
L - Illumina 1.8+ Phred+33, scores des séquences brutes compris entre 0 et 41
Séquençage par espace colorimétrique
Concernant les données de séquençage issues de la technique SOLiD, l'ADN est séquencé par une méthode d'espace colorimétrique, excepté pour la première position. Les scores de qualité sont ceux du variant Sanger. Les programmes de traitement de séquences diffèrent dans leur préférence concernant le score de qualité associé à la première position : certains incluent un score de qualité pour cette position (défini à 0 donc avec le caractère "!"), d'autres non. Les Archives de séquences incluent automatiquement un score de qualité à cette position"
Conversion de variants et de formats
Conversion de variants FASTQ
Du fait de l'incompatibilité entre les variants du format FASTQ, il est nécessaire de les convertir dans l'un des variants (de préférence le variant Sanger) pour pouvoir utiliser ensemble des fichiers d'origine différentes. Pour cela des convertisseurs ont été intégrés dans les différents projets soutenus par l'Open Bioinformatics Foundation :
- BioPython version 1.51 et suivantes (conversion variants Sanger, Solexa et Illumina 1.3+)
- EMBOSS version 6.1.0 patch 1 et suivantes (conversion variants Sanger, Solexa et Illumina 1.3+)
- BioPerl version 1.6.1 et suivantes (conversion variants Sanger, Solexa et Illumina 1.3+)
- BioRuby version 1.4.0 et suivantes (conversion variants Sanger, Solexa et Illumina 1.3+)
- BioJava version 1.7.1 to 1.8.x (conversion variants Sanger, Solexa et Illumina 1.3+)
D'autres outils existent permettant également ces conversions :
- MAQ peut convertir des fichiers du variant Solexa vers le variant Sanger (utiliser ce patch pour prendre en compte les fichiers Illumina 1.3+)
- Fastx toolkit incluant son programme fastq_quality_converter peut convertir les variants Illumina au variant Sanger
Conversion de format FASTQ vers FASTA
Sous un système UNIX, il est possible de convertir aisément un ficher du format FASTQ vers un format FASTA en utilisant la ligne de commande suivante :
cat input_file.fastq | paste - - - - | awk '{print ">"$1"\n"$2}' > output_file.fa
ou si le fichier est compressé avec gunzip :
zcat input_file.fastq.gz | paste - - - - | awk '{print ">"$1"\n"$2}' > output_file.fa
Cette commande ne conserve du fichier FASTQ que les lignes contenant l'identifiant (avec la description si présente) et les séquences associées. Le caractère "@" introduisant la ligne de l'identifiant est également remplacé par ">".
Références
- (en) Cock PJ., Fields CJ., Goto N., Heuer ML. & Rice PM., « The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. », Nucleic Acids Research, vol. 38, no 6, , p. 1767-71 (ISSN 1362-4962, PMID 20015970, DOI 10.1093/nar/gkp1137)
- (en) Sequencing Analysis Software User Guide: For Pipeline Version 1.4 and CASAVA Version 1.0, (lire en ligne)
- (en) Sequencing Analysis Software User Guide: For Pipeline Version 1.5 and CASAVA Version 1.0, (lire en ligne)
- (en) Sequence/Alignment Map format Version 1.0, (lire en ligne)
- (en) skruglyak, « Upcoming changes in CASAVA », sur Seqanswers.com, (consulté le )
- (en) Bio.X2Y, « Illumina FASTQ Quality Scores - Missing Value », sur Seqanswers.com (consulté le )
- (en) Using Genome Analyzer Sequencing Control Software, Version 2.6, Catalog # SY-960-2601, Part # 15009921 Rev. A, (lire en ligne)
- (en) « Site internet du projet SolexaQA » (consulté le )
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « FASTQ format » (voir la liste des auteurs).
Voir aussi
Articles connexes
Liens externes
- (en)MAQ page web discutant des divers variants FASTQ
- (en)Outils de traitement de fichiers FASTQ sur Galaxy
- (en)Fastx toolkit : ensemble de programmes en ligne de commande pour le prétraitement des séquences courtes de fichiers FASTA/FASTQ
- (en)Fastqc : outil de contrôle qualité pour les données de séquençage haut-débit
- (en)PRINSEQ peut être utilisé pour le contrôle qualité et le filtrage, reformatage ou le nettoyage de données de séquences (versions web et en ligne de commande )