Déficit immunitaire combiné sévère lié à l'X
Le déficit immunitaire combiné sévère lié à l'X est une immunité humorale et cellulaire déficitaire par absence de lymphocyte T, et de cellules NK. D'un point de vue nosologique, il s'agit d'un déficit immunitaire héréditaire de type déficit combiné sévère[1]. Cette maladie génétique ne touche que les garçons et les premiers signes apparaissent entre trois et six mois de vie.
Autres noms
- Immunodéficience combinée sévère T- B+ due à un déficit en chaine gamma (γ).
- Déficience de la chaîne commune γ des récepteurs des lymphocytes T
Description
Ce déficit se caractérise par des infections bactériennes ou virales sévères et récurrentes. Les signes comprennent un défaut de croissance, une absence d'amygdale, des candidoses buccales, et l'absence de guérison malgré un traitement bien conduit. Elles débutent dès les premiers mois de vie et sont généralement associées à une diarrhée et à un retard staturopondéral. Le malade est rendu fragile à toutes les infections, car son organisme se défend mal.
Cause
Cette maladie est liée à une (des) mutation(s) du gène codant la sous-unité γc[2], protéine transmembranaire commune à plusieurs récepteurs d’interleukines (Il2, Il4, Il7, Il9, Il15 et Il21). Ces mutations conduisent à un défaut d’expression ou de fonctionnalité de la protéine γc à la surface des précurseurs lymphocytaires. Ceci provoque un blocage précoce de la différenciation de ces précurseurs en lymphocytes T et en cellules NK. Les NK sont capables de lyser des cellules malades sans nécessiter d’activation préalable et sans entrer en contact avec le pathogène. Les lymphocytes T eux ont un rôle dans la réponse immunitaire secondaire, c'est-à-dire la réponse de l’immunité cellulaire[3].
Incidence et prévalence
La prévalence de cette maladie rare est de 1 à 9⁄100000.
L’incidence est d’environ 1⁄200000 naissances.
Dépistage néonatal
En France, depuis 2022, la Haute Autorité de Santé (HAS) recommande le dépistage néonatal des déficits immunitaires combinés sévères, dont celui lié à l'X[4].
Diagnostic
Il existe deux façons de diagnostiquer ce déficit :
- un diagnostic par analyse de la population lymphocytaire.
- un diagnostic anténatal, réalisé à 11 semaines d’aménorrhée. Si dans la famille il y a déjà eu ce type de déficit, la détection de la mutation est directe. Une biopsie de trophoblaste est alors réalisée. Dans tous les cas, et surtout s’il y a un doute sur la transmission à l’enfant du déficit, il est fait une étude de la population lymphocytaire. L’étude immunologique est réalisée à 20 semaines de grossesse sur ponction fœtale.
L’étude de la population lymphocytaire va révéler une lymphopénie pour les lymphocytes T (CD3+) aussi bien CD4+ que CD8+, et les cellules NK, alors que les lymphocytes B sont présents en nombre normal voire ont augmenté. Ce déficit lymphocytaire conduit à une agammaglobulinémie. On obtient alors ce phénotype clinique et immunologique, DICS T-, B+ NK-[5].
Traitement et prise en charge
En l’absence de traitement, la pathologie est fatale dans la première année de vie, de par la survenue d’infections sévères et récurrentes.
Il existe trois traitements possibles :
- un traitement de substitution
- une greffe de moelle osseuse
- la thérapie génique
Traitement de substitution
Le traitement de substitution est une injection d’immunoglobulines par voie intraveineuse. Les immunoglobulines ne permettent pas de rétablir la fonction des lymphocytes T déficients, mais elles sont bénéfiques puisqu’elles apportent les anticorps manquants à cause des lymphocytes T déficients. Ces injections doivent être répétées et ne constituent qu'une solution d'attente.
Greffe de moelle osseuse
La greffe de moelle osseuse est le traitement de référence[6]. C’est la seule option curative pour la plupart des enfants affectés, et est d'autant plus efficace qu'elle est réalisée précocement[7].
Il y en a deux types :
- La greffe allogénique, possédant le même antigène tissulaire d’histocompatibilité (HLA), provenant d’un donneur intrafamilial. Malheureusement, moins de 20 % des patients atteints possèdent un tel donneur.
- La greffe de moelle osseuse provenant d’un donneur haplo-identique. Cependant, un tel traitement n’est pas sans risques à court et à long terme : maladie du greffon contre l’hôte (GVHD), infections persistantes dues à une restauration incomplète de l’immunité, déficit immunitaire B résiduel avec obligation d’un traitement continu par immunoglobulines, déclin à moyen ou long terme des cellules T fonctionnelles.
La première greffe de moelle osseuse pour DICS a été effectuée en 1968 et le patient est encore en vie à ce jour.
Thérapie génique
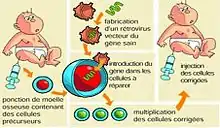
Les patients atteints du syndrome de déficit immunitaire combiné sévère lié à l’X pourraient aussi être traités par thérapie génique. Cette thérapie consiste au transfert d’un gène sain dans le génome d’une cellule mutante, afin qu’il permette la production de la protéine manquante. Elle consiste à :
- la fabrication, par isolement et clonage, d’un gène thérapeutique ;
- l’introduction dans des cellules souches du patient de ce gène ex vivo ;
- la réinjection de ces cellules au patient qui va pouvoir développer une immunité.
Cette thérapie n'en est encore qu’à l’état d’essais. Elle fut mise au point par les Français Alain Fischer et Marina Cavazzana-Calvo en 1993. Les premiers essais cliniques, réalisés en 2000, furent un succès et rétablirent une immunité chez quasiment tous les sujets traités. Mais l'insertion du gène, bien que rendant leur immunité aux bébés, peut également déclencher des cancers, et sur les neuf patients traités pendant le premier essai clinique français, quatre développèrent une leucémie, et un en mourut[8]. Pour plus de sécurité les essais ont donc dû être suspendus, afin de comprendre ce qui avait pu se produire. En 2010, de nouveaux essais cliniques ont pu reprendre chez neuf enfants avec un vecteur modifié, sans développement secondaire de leucémie[9]. L'association d'une thérapie génique à du busulfan permet d'en améliorer les résultats[10].
La prise en charge des patients se fait dans une unité spécialisée en immunologie et en hématologie. Il faut prohiber pour ces personnes l’utilisation de vaccins vivants, ainsi que la transfusion de produits sanguins non irradiés.
Mode de transmission
Transmission héréditaire récessive. Le gène codant la protéine est localisé sur le chromosome X, et l’allèle muté est récessif. Les garçons expriment donc la maladie et les filles peuvent être porteuses saines à moins qu’elles possèdent deux chromosomes X mutés.
Notes et références
- A. Bousfiha et al., « Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification », Journal of Clinical Immunology, (lire en ligne)
- Noguchi M, Yi H, Rosenblatt HM et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans, Cell, 1993;73:147-157
- http://www.afssaps.fr/Infos-de-securite/Information-produit-Information-traitement/Deficit-immunitaire-combine-severe-lie-a-l-X-DICS-X/(language)/fre-FR
- « La HAS propose l’extension du dépistage néonatal au déficit immunitaire combiné sévère (DICS) », (consulté le )
- http://www.orpha.net/data/patho/FR/fr-xdics.pdf
- Buckley RH, Schiff SE, Schiff RI et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency, N Engl J Med, 1999;340:508-516
- Myers LA, Patel DD, Puck JM, Buckley RH, Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival, Blood, 2002;99:872-878
- (en) Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, Martinache C, Rieux-Laucat F, Latour S, Belohradsky BH, Leiva L, Sorensen R, Debré M, Casanova JL, Blanche S, Durandy A, Bushman FD, Fischer A, Cavazzana-Calvo M, « Efficacy of gene therapy for X-linked severe combined immunodeficiency », N Engl J Med, vol. 363, no 4, , p. 355-64. (PMID 20660403, PMCID PMC2957288, DOI 10.1056/NEJMoa1000164, lire en ligne [html], consulté le )
- Hacein-Bey-Abina S, Pai SY, Gaspar HR et al. A modified γ-Retrovirus vector for X-Linked severe combined immunodeficiency, N Engl J Med, 2014; 371:1407-1417
- Mamcarz E, Zhou S, Lockey T et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1, N Engl J Med, 2019;380:1525-1534 ,
Voir aussi