Chromatographie à échange d'ions
La chromatographie par échange d’ions (IC, Ion-exchange Chromatography), souvent appelée chromatographie ionique, désigne l’identification des ions à l’aide de résines échangeuses d’ions[1]. Cette technique appartient aux méthodes chromatographiques qui sont largement utilisées dans tous les domaines de l’analyse chimique pour leur souplesse d’utilisation et la diversité de leurs applications[2].

La chromatographie par échange d'ions sépare les molécules selon leurs groupes chargés respectifs. Les ions de l’analyte subissent des interactions ioniques avec des charges opposées fixées sur la phase stationnaire, ce qui entraîne leur rétention. La phase stationnaire en question est constituée d'une matrice immobile qui contient des groupes fonctionnels chargés[3].
Le développement de la technique a commencé au milieu des années 1970, lorsque l’on a découvert que des mélanges d’anions et de cations pouvaient être séparés à l’aide de colonnes CLHP (Chromatographie en phase liquide à haute performance) constituées de résines échangeuses de cations ou d’anions. A cette époque, la détection des ions était en général effectuée par des dosages conductimétriques[1].
Historique
À la fin des années 1960 débute le développement de la chromatographie ionique, mais application à la séparation des espèces ioniques retardée par manque d’une technique générale et sensible[1].
En 1975, La chromatographie ionique est introduite par Small, Stevens et Bauman comme nouvelle méthode analytique[4].
Elle évolue rapidement dans sa détection pour aller vers une détection plus sensible des ions par leur conductance électrique. Une colonne dite suppresseur réduit chimiquement la conductance de l’éluant[4].
En 1979, Fritz et al. décrit une séparation et une détection alternative à celle de la cellule de conductivité classique. Le principe est d’utiliser des résines échangeuses d’ions de faible capacité et des éluants à faibles teneur en ions (permettant une conductance faible)[5].
À la fin des années 1970, des techniques chromatographiques ioniques ont été utilisées pour analyser les ions organiques pour la première fois[6].
En 1983 : Développement d’un détecteur ampérométrique pulsé permettant une détection très sensible des hydrates de carbone, des acides aminés et des composés soufrés divalents[7] - [8].
Dans le début des années 1990, le développement des colonnes avec des sélectivités spéciales a été effectué[6].
Principe
Ce procédé de séparation est basé sur des processus d'échange d'ions se produisant entre les analytes dans la phase mobile et la résine fixée sur la colonne. La chromatographie d'échange d'ions est utilisée pour séparer les anions et les cations inorganiques et organiques. La séparation des anions est réalisée avec une colonne d’échange anionique et la séparation des cations est réalisée avec une colonne d’échange cationique[6].
Échange anionique
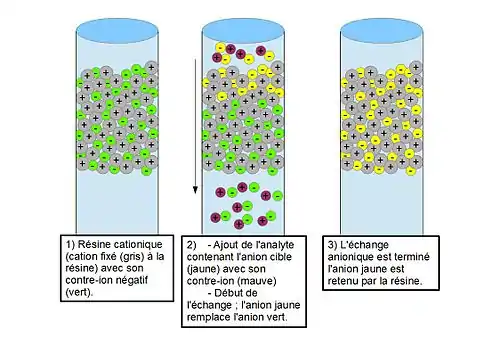
Lors d’un échange anionique, une résine cationique est utilisée (étape n°1). L’ajout de l’analyte provoque le remplacement du contre-ion de la résine par celui de l’ion soluté (étape 2). Lorsque le contre-ion du site d'échange d'ions est remplacé par un ion soluté (étape 3), celui-ci est temporairement retenu par la charge fixe. Les divers ions analytes restent pendant une période différente dans la colonne en raison de leurs affinités différentes vis-à-vis de la phase stationnaire, et ainsi, la séparation est provoquée. Si un échantillon avec les anions A− et B− est injecté dans la colonne, on échange ces anions par des anions compétiteurs de la colonne selon le processus d'équilibre réversible donné par les équations suivantes[6] :
Résine-N+R3 E− + A− k1 ⇌ Résine- N+R3A− + E−
Résine-N+R3 E− + B− k2 ⇌ Résine- N+R3B− + E−
La séparation des anions est déterminée par leurs différentes affinités avec la phase stationnaire. La constante déterminant le processus d'équilibre est le coefficient de sélectivité K, qui est défini comme suivant[6] :
![{\displaystyle K={\frac {[X^{-}]_{s}*[E^{-}]_{m}}{[E^{-}]_{s}*[X^{-}]_{m}}}}](https://img.franco.wiki/i/45b87c26bfb4ee0f6a339e77eb750e1da03f9f97.svg)
[X−]m.s : Concentration des ions échantillons dans la phase mobile (m) ou dans la phase stationnaire (s).
[E−]m.s : Concentration en bicarbonate dans la phase mobile (m) ou dans la phase stationnaire (s).
Échange cationique
La réaction d'échange avec un cation M+ qui se produit sur la phase stationnaire d'un échangeur cationique peut être exprimée comme suivant :
Résine-SO3−E+ + M+ à Résine-SO3−M+ + E+
La séparation des cations est déterminée par leurs différentes affinités avec la phase stationnaire. La constante de sélectivité K est déterminée de la même manière que pour l’échange anionique[6].
Matériel et montage

Principe général
Pour effectuer une séparation, l'éluant est tout d'abord pompé seul à travers le système jusqu'à ce que la ligne de base apparaisse stable à l’écran (quelques minutes à quelques heures selon le type de résine et d’éluant utilisé)[6] - [9]. L’échantillon est ensuite introduit, soit manuellement soit avec un échantillonneur automatique, dans une boucle d’injection de volume connu[6] - [9]. Une solution aqueuse tamponnée, appelée phase mobile, emmène l'échantillon de la boucle vers la colonne[6] - [9]. Celle-ci est composée d’une phase stationnaire constituée d’une résine contenant des groupes fonctionnels chargés[9]. Les analytes cibles (anions ou cations) sont retenus sur la phase stationnaire, mais peuvent être détachés de celle-ci en ajoutant une espèce de charge similaire dans l’éluant. Par exemple, dans la chromatographie d'échange cationique, l'analyte chargé positivement peut être déplacé en ajoutant des cations sodium (éluant). Les analytes cibles doivent alors être détectés par différents moyens, typiquement par la conductivité ou par absorbance UV / visible[9].
L'éluant
L’éluant porte l’échantillon de la boucle d’injection jusqu’à la colonne[9].
L'éluant utilisé dans la chromatographie anionique contient un anion éluant, E−. Habituellement Na+ ou H+ sera le cation associé à celui-ci[9].
L'anion éluant doit être compatible avec la méthode de détection utilisée[6] - [9]. Pour la conductivité, la détection de E− doit avoir, soit une conductivité significativement plus faible que les ions de l'échantillon, soit être capable d'être convertie en une forme non ionique par un système de suppression chimique. Lorsqu'on utilise une détection spectrophotométrique, E− sera souvent choisi pour sa capacité à ne pas absorber dans la région UV ou visible[9].
Dans une chromatographie cationique, l’éluant contient un cation E+. Ce cation doit également être compatible avec la méthode de détection utilisée[9].
Dégazage de l'éluant
Le dégazage de l'éluant est important car si l'air entre dans la pompe, il peut causer sa perte. Habituellement, de l'eau dégazée est utilisée. Enfin, il est préférable de changer régulièrement les éluants pour garder leur concentration précise[9].
La pompe
Le but de la pompe est de délivrer l’éluant dans la colonne à un débit fixe, qui est généralement de 1mL/min[9].
Pour provoquer l’aspiration, le piston est retiré de la tête de pompe. Ensuite, ce même piston retourne dans celle-ci. L'augmentation de pression permettra alors d’ouvrir la soupape de sortie, forçant l’éluant à s’écouler vers la soupape d'injection puis à travers la colonne[9].
Il est primordial de purger la pompe afin d’éviter que de l’air n'entre dans celle-ci et ne cause sa défaillance[9].
L'injecteur
L’injecteur permet d’introduire l’échantillon dans le chromatographe. Le système d'injection peut être manuel ou automatisé, mais tous deux reposent sur une soupape d'injection. Celle-ci est conçue pour introduire l'échantillon dans la boucle d’injection[6] - [9].
La variation de volume est généralement inférieure à 0,5 % de précision d’une injection à une autre[9]. La boucle peut être partiellement remplie (injection de boucle partielle) ou complètement remplie (injection de boucle complète). Si une injection en boucle partielle est utilisée, la boucle ne doit pas être remplie à plus de 50 % de son volume total sinon l'injection pourrait ne pas être précise. Dans l'injection de boucle complète, l'échantillon est poussé complètement à travers la boucle[9]. La taille de la boucle est comprise entre 5 et 200 μL[6] - [9]. Normalement, on utilise au moins le double du volume d’échantillon voulu pour remplir la boucle afin de s’assurer que celle-ci soit bien pleine. L’excès d’échantillon part ensuite au déchet[9].
L'injection de l'échantillon est réalisée en tournant la soupape ce qui entraîne le contenu de la boucle d'injection dans le courant d'éluant[9].
Les vannes d'injection nécessitent un entretien périodique et doivent généralement être entretenues après environ 5 000 injections[9].
La colonne
Les anions sont séparés dans une colonne composée d’une résine échangeuse d'anions. De même, les cations sont séparés dans une colonne contenant une résine échangeuse de cations. Les principes de séparation des anions et des cations sont décrits dans le principe.
Les colonnes IC sont généralement faites de PEEK (PolyEtherEtherKetone, PolyEtherEtherCetone). La taille de colonne typique utilisée en chromatographie ionique est de 150 × 4,6 mm bien que des colonnes comprises entre 10 et 250 mm[6] - [9] avec un diamètre de particules de 5 ou 10 μm soient également utilisées[9].
La séparation est basée sur les différences dans l'équilibre d'échange des différents ions analytes avec la résine[6]. Ainsi, si l'ion analyte A1− a une affinité inférieure pour la résine à celle de l'ion A2−, alors A1− se déplacera à un rythme plus rapide à travers la colonne[9].
La colonne peut être contenue dans un four pour contrôler les conditions et optimiser les séparations[9].
Les résines
Les échangeurs d'anions peuvent être constitués de polymères tels que du polystyrène, de l’éthylvinylbenzène ou du métacrilylate copolymérisés avec le divinylbenzène[6] ou de silice fonctionnalisés avec des groupes échangeurs d’ions. Bien que de nombreux types d'échangeurs d'anions soient disponibles pour la chromatographie, le type "base forte" avec des groupes fonctionnels d'ammonium quaternaire est le plus courant. Les contre-ions sont principalement des groupes chlorure[9].
Les groupes ammonium quaternaire sont chimiquement liés aux résines tandis que les groupes chlorure sont capables de subir un échange d'ions avec d'autres anions[9].
Un échangeur de cations sera généralement fonctionnalisé avec des groupes chargés négativement tels que des groupes sulfonates, carboxylates ou phosphonates[6].
Détecteur et suppresseur
Le détecteur le plus courant et le plus utilisé pour la chromatographie ionique est le détecteur de conductivité. Cependant, les détecteurs spectrophotométriques et autres (UV/vis, fluorescence, ampérométrie…[6]) peuvent également être très utiles[9].
Détecteur de conductivité
La conductivité est la capacité d'une solution contenant un sel à conduire de l'électricité à travers deux électrodes[9]. La capacité de la solution à conduire est directement proportionnelle à la teneur en sel et à la mobilité des anions et des cations[9]. Les substances moléculaires telles que les solvants (eau, méthanol) n'entraînent pas l'électricité et ne sont donc pas détectées par le détecteur[9].
En raison de sa sensibilité élevée et de sa capacité à réduire le bruit de fond, la forme la plus commune de détection de conductivité est avec l'utilisation d’un suppresseur. Le suppresseur chimique fournit un moyen simple de réduire la conductance de fond de l’éluant et, en même temps, d’améliorer la conductance des ions analytes[6] - [9]. Après la colonne, une seconde colonne d’échange d’ions est placée, il s’agit du suppresseur. Pour l’analyse anionique, l’ion de l’éluant est anionique, c’est donc une colonne d’échange de cations H+ qui est utilisée. Les réactions de suppression anionique sont les suivantes[9] :
Éluant :
NaOH (Forte conductivité) + RSO3H à H2O (faible conductivité) + RSO3Na
Analyte :
NaX (conductivite moyenne) + RSO3H à HX (forte conductivité) + RSO3Na
L'éluant de base (OH−) est neutralisé par H+ de l'échangeur de cations (suppresseur). Le contre-ion de l’analyte (Na+ X−) est converti en contre-ion hydrogène plus conducteur (H+ X−). La conductance de l'éluant entrant dans le détecteur est donc très faible car pratiquement tous ses ions ont été éliminés par l'unité de suppression[9].
Le détecteur est connecté à un dispositif d'acquisition de données de sorte qu'un diagramme de séparation (signal en fonction du temps) peut être tracé automatiquement. La lecture de la conductance G est donnée en Siemens (S), ou plutôt en microSiemens (μS) car les solutions sont généralement diluées[9].
Cependant, un acide faible avec un pKa supérieur à environ 6 ne pourra pas être détecté avec un détecteur de conductivité muni d’un suppresseur parce que tous les anions seraient convertis en acide par celui-ci. Pour la détection de sels d'acides faibles, il faut des détections par conductivité sans suppresseur[9].
Ainsi, la détection directe d’ions est également possible par chromatographie ionique à colonne unique.
Son fonctionnement est basé sur la mesure des petites différences de conductivités qui existent entre les ions cibles et les ions de l’éluant. Pour amplifier ces différences, on utilise des échangeurs à faible capacité, ce qui permet d’utiliser des solutions diluées d’éluant. De plus, on choisit des éluants qui ont une faible conductivité[10].
La chromatographie ionique à colonne unique offre l’avantage de ne pas nécessiter d’équipement spécial de suppression. Toutefois, dans le cas du dosage des anions, cette méthode est moins sensible que celle utilisant une colonne de neutralisation[10].
Système Data et interprétation des résultats
Les résultats de la séparation chromatographique sont généralement affichés sur un ordinateur. Le chromatogramme obtenu indique le signal en fonction du temps. Les temps de rétention sont utilisés pour confirmer l'identité du pic inconnu par rapport à un étalon. La surface du pic inconnu est généralement comparée à une droite d’étalonnage préalablement réalisée avec des étalons de concentrations connues[9].
Applications
La chromatographie ionique convient pour l'analyse des anions et des cations qu’ils soient inorganiques ou organiques. Le grand nombre d'applications citées ci-dessous illustre la polyvalence de cette méthode[11].
La chromatographie ionique est devenue un outil indispensable pour le chimiste analytique, en particulier dans le domaine de l'analyse anionique[11].
Dans de nombreux cas, cette méthode a remplacé les méthodes chimiques humides conventionnelles telles que la titration, la gravimétrie ou encore la colorimétrie, qui sont toutes laborieuses, fastidieuses et parfois sujettes à des interférences[11].
Dans le domaine de l'analyse des cations, elle est attrayante en raison de sa détection et de sa sensibilité. Elle fournit un complément bienvenu aux méthodes de spectrométrie atomique telles que AAS (Spectroscopie d’absorption atomique) et ICP (spectrométrie à plasma à couplage inductif)[11].
Les applications de chromatographie ionique sont principalement dans les domaines suivants : analyse environnementale (air, sol et eau), chimie des centrales électriques, industrie des semi-conducteurs, aliments et boissons, galvanoplastie, produits pharmaceutiques, biotechnologie, ou encore l’agriculture[1] - [11].
Notes et références
- Skoog, Holler, Nieman, Principes d’analyse instrumentale, Bruxelles, De Boeck, 2003, 919 p..
- J-J. Minet, V. Eudes, C. Costanza, X. Archer, M-B. Le Borgne, G. Baron, (2010), L’utilisation des méthodes chromatographiques en police scientifique, L’actualité chimique no 342-343, p. 62-69.
- Jungbauer, Alois,, Hahn, Rainer (2009), "Chapter 22 Ion exchange chromatography" Guide to protein purification, 2nd edition. Methode in enzymology. 463. pp 349-371
- H. Small, T.S Stevens, W.S Bauman (1975), Novel ion exchange chromatographic method using conductimetric detection, Analytical Chemistry. Vol. 47 no 11, p. 1801
- D.T Gjerde, J.S Fritz, G Schmuckler (1979), Anion chromatography with low conductivity eluents : J. Chromatogr. Vol. 186 p. 509
- Joachim Weiss (2004) Handbook of Ion Chromatography Volume 1, Weinheim : WILEY-VCH, 547p
- R.D Rocklin, C.A Pohl, (1983), Determination of Carbonylates by anion exchange chromatography with pulsed amperometric detection J. Liq. Chromatogr. Vol. 6 p. 1577
- D.A Martens, W.T Frankenberger (1992), Pulsed Amperometric detection of amino acids separated by anion exchange chromatography J. Liq. Chromatogr. Vol. 15, p. 423
- James S. Fritz, Douglas T. Gjerde, (2000) Ion Chromatography, Weinheim : WILEY-VCH, 247p
- Skoog, West, Holler, Crouch, (2015) Chimie analytique. Bruxelles : De Boeck, 1049p
- Joachim Weiss (2004) Handbook of Ion Chromatography Volume 2, Weinheim : WILEY-VCH, p. 549-839