Syndrome d'Apert
Le syndrome d’Apert est une craniosynostose en rapport avec une mutation du gène FGFR2. Cette mutation du gène FGFR2 est responsable d’autres craniosynostose regroupées sous le nom de craniosynostose FGFR dépendante.
| Spécialité | Génétique médicale |
|---|
| CISP-2 | A90 |
|---|---|
| CIM-10 | Q87.0 |
| CIM-9 | 755.55 |
| OMIM | 101200 |
| DiseasesDB | 33968 |
| MedlinePlus | 001581 |
| eMedicine | 941723 |
| MeSH | D000168 |
![]() Mise en garde médicale
Mise en garde médicale
Les sutures du crâne qui fusionnent dans cette maladie sont les sutures coronales.
Le syndrome d'Apert est désigné ainsi en raison de sa découverte par le médecin français Eugène Apert, qui le premier a décrit le syndrome en 1906[1].
Autres noms de la maladie
- Acrocephalosyndactylie type I
- Maladie d’Apert-Crouzon
- Acrocéphalosyndactylie type II
- Céphalodactylie de Vogt
Étiologie
Mutation du gène FGFR2, ou fibroblast growth factor receptor-2 localisé sur le locus q26 du chromosome 10. Il existe deux allèles de cette mutation. Certaines études suggèrent que l’âge paternel augmenterait le risque d'apparition de cette pathologie. Le syndrome d'Apert est réputé s'accompagner fréquemment d'une déficience intellectuelle, à l'origine de laquelle des malformations cérébrales pourraient jouer un rôle important.
Incidence
Un nouveau-né sur 100 000 en France et 1 nouveau-né sur 50 000 aux États-Unis.
Description

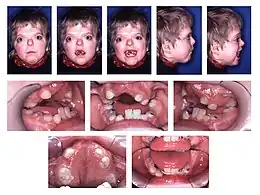
- Le diagnostic clinique se fait sur la présence de fusion prématurée des sutures coronales avec des caractéristiques du visage. La croissance des os du crâne se faisant perpendiculairement aux sutures, le crâne prend une forme de tour.
- Des anomalies cérébrales sont possibles telles que l’agénésie du corps calleux.
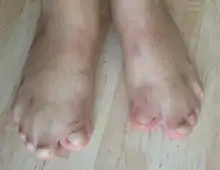
- Il existe des anomalies avec une fusion des doigts des mains et des pieds, ou syndactylie. Cette syndactylie comprend 4 doigts sur 5 réalisant l’aspect dit « en cuiller », « en main d’accoucheur » ou « en moufles ».
- Le retard mental est fréquent et important.
- Troubles du langage possibles.
Diagnostic
Anténatal
- Possible par échographie soit par visualisation de la syndactylie ou par la déformation du crâne en forme de tour (turricéphalie). L’échographie en 3D peut être très utile pour mettre en évidence la fusion prématurée des sutures coronales.. L’imagerie à résonance magnétique est indispensable pour compléter l’étude du cerveau du fœtus.
Post natal
Les caractéristiques cliniques font facilement le diagnostic.
Différentiel
Est celui d’une craniosynostose.
Mode transmission
La plupart des cas sont des mutations de novo mais les cas familiaux suggèrent une transmission autosomique dominante.
Traitement
Une investigation soigneuse comprenant un examen en résonance magnétique nucléaire est nécessaire pour rechercher des malformations cérébrales associées. Les patients doivent être opérés tôt, idéalement avant l'âge de 9 mois. La qualité de l'environnement sociofamilial doit être optimisée[2].
Conseil génétique
Une enquête familiale est indispensable. Le diagnostic de la maladie est possible par étude du caryotype en début de grossesse.
Voir aussi
Notes et références
- (en) « Article « Syndrome d'Apert » », sur Who Named It?
- D Renier, E Arnaud, G Cinalli, D Marchac, L Brunet, G Sebag, C Sainte-Rose, M Zerah « Pronostic mental du syndrome d'Apert » Archives de Pédiatrie 1996;3(8):752-60. DOI 10.1016/0929-693X(96)82156-7, Éditions scientifiques et médicales Elsevier