Syndrome MELAS
Le syndrome MELAS est une maladie mitochondriale qui débute typiquement dans l'enfance se manifestant essentiellement par un syndrome neurologique. Le terme « MELAS » est une abréviation de l'anglais « mitochondrial encephalopathy with lactic acidosis and stroke-like episodes » : encéphalopathie mitochondriale, acidose lactique et épisodes déficitaires neurologiques.
Description
Le développement précoce psychomoteur est normal mais l'enfant est habituellement de petite taille. L'âge de début des symptômes est entre deux et dix ans, comprenant habituellement des crises d'épilepsie tonico-clonique, des vomissements, des céphalées et une anorexie. Une fatigabilité excessive à l'exercice ou des faiblesses musculaires peuvent aussi être les premières manifestations. Il peut exister une surdité.
Les convulsions sont souvent associées à des hémiparésies transitoires, des déficits moteurs ou des épisodes de cécité corticale. Les déficits moteurs peuvent être associés à des troubles de la conscience et sont souvent récurrents. L'effet cumulatif de ces épisodes de déficit moteur aboutit graduellement à une perte de l'autonomie, une perte de la vision et une démence au début de l'âge adulte. La surdité est habituelle. Il peut exister un diabète[1].
Il existe une atteinte cardiaque dans un tiers des cas[2] avec des présentations variables[3].
Le tableau est toutefois extrêmement variable pour la même mutation[1].
Autres noms
- Myopathie mitochondriale - encéphalopathie - acidose lactique
Causes
Le syndrome est causé par des mutations de l'ADN mitochondrial. Plusieurs gènes peuvent être en cause dont l' ARNt-Leucine. la mutation la plus fréquemment associée est la mutation A3243G dans l'ARNtLeu de l'ADN mitochondrial.
Les manifestations sont variables suivant le nombre de mitochondries atteint[4].
Diagnostic
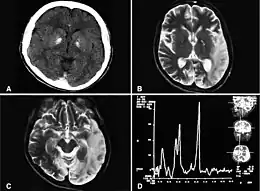
les lésions cérébrales ne sont pas de topographie vasculaire et sont plus souvent postérieures qu'antérieures[5].
Traitement et prise en charge
En cas d'anesthésie du patient, des précautions particulières sont à pendre car les myopathies mitochondriales induisent souvent une sensibilité accrue aux myorelaxants utilisés par les anesthésistes[6].
Traitement
Il est uniquement symptomatique. Beaucoup de patients prennent des suppléments nutritionnels pour tenter d'augmenter le métabolisme (Riboflavine, L-carnitine, coenzyme Q10) mais le niveau de preuve de l'efficacité reste faible[7]. L'arginine semble toutefois améliorer les déficits neurologiques[8].
Conseil génétique
Mode de transmission
Hérédité mitochondriale, rares mutations de novo, le diagnostic pré natal est possible mais difficile du fait de l'hétéroplasmie. La mutation 3243G>A est la plus fréquente(80%)[9].
La transmission peut être maternelle (par les mitochondries maternelles) mais aussi de type autosomique, le plus souvent récessive par l'ADN autosomale codant pour des protéines mitochodriales.
Notes et références
- (en) Mancuso M, Orsucci D, Angelini C et al. « The m.3243A>G mitochondrial DNA mutation and related phenotypes: a matter of gender? » J Neurol. 2014;261:504-10.
- Hsu HR, Yogasundaram H, Parajuli N, Valtuille L, Sergi C, Oudit CY, MELAS syndrome and cardiomyopathy: linking mitochondrial function to heart failure pathogenesis, Heart Fail Rev, 2016;21:103-116
- Niedermayr K, Pölzl G, Scholl-Bürgi S et al. Mitochondrial DNA mutation "m.3243A>G"-Heterogeneous clinical picture for cardiologists ("m.3243A>G": A phenotypic chameleon), Congenit Heart Dis, 2018;13:671-677
- McMillan RP, Stewart S, Budnick JA et al. Quantitative variation in m.3243A > G mutation produce discrete changes in energy metabolism, Sci Rep, 2019;9:5752
- Murakami H, Ono K et al. MELAS: mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes, Brain Nerve, 2017;69:111-117
- (en) Thompson VA, Wahr JA. « Anesthetic considerations in patients presenting with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome » Anesth Analg. 1997; 85:1404–6.
- (en) Karaa A, Kriger J, Grier J et al. « Mitochondrial disease patients’ perception of dietary supplements’ use » Mol Genet Metab. 2016;119:100-8.
- (en) Koenig MK, Emrick L, Karaa A et al. « Recommendations for the management of strokelike episodes in patients with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes » JAMA Neurol. 2016;73:591-594
- « Orphanet: MELAS », sur www.orpha.net (consulté le )
Voir aussi
Article connexe
Liens externes
- Page spécifique sur Orphanet
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 540000
- (en) Salvatore DiMauro, MELAS In : GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997-2005. .